INTRODUCTION
Aging causes progressive impairment of homeostasis in the human body. In general, aging is affected by environmental changes and can lead to various diseases (Kirkwood, 2005). In particular, the natural process of aging is among the most major risk factors that increase susceptibility to develop cardiovascular diseases (Dai et al., 2012), which often feature left ventricular hypertrophy (Dai and Rabinovitch, 2009), diastolic dysfunction (Bursi et al., 2006), valve degeneration (Karavidas et al., 2010), increased myocardial fibrosis (Olsen et al., 2005), and increased atrial fibrillation (Gerhard-Herman et al., 2017).
Mitochondria play an important role in maintaining a healthy heart and regulating various cellular processes, including adenosine triphosphate (ATP), which is utilized as energy in the heart through oxidative phosphorylation (OXPHOS) (Chaudhary et al., 2011). The free radical theory of aging proposed 60 years ago posits that the activity of reactive oxygen species (ROS) is the important determinant of lifespan and induction of cardiac diseases (Harman, 1956). In general, ROS are generated by multiple enzymes and organelles in the cell, such as NADPH oxidase at the plasma membrane, lipid oxidation in peroxisomes, and especially OXPHOS in mitochondria (Dai et al., 2012). Appropriate levels of mitochondrial H2O2 emission, which is regarded as a physiological ROS marker, are essential in regulating a cellular redox environment of cells and mitochondrial function (Jones, 2008). However, excessive ROS emissions, such as H2O2, can induce mitochondrial and cellular dysfunctions, which lead to impaired mitochondrial DNA (Martindale and Holbrook, 2002), causing senescence in many cell types (Ben-Porath and Weinberg, 2005). Excessive mitochondrial H2O2 emission has also been related to aging (Bejma et al., 2000; Kuka et al., 2013; Petrosillo et al., 2009).
The Ca2+ signals in mitochondria regulate key cellular functions, such as energy production, by regulating mitochondrial enzymes like pyruvate dehydrogenase, isocitrate dehydrogenase, and alpha-ketoglutarate dehydrogenase in the tricarboxylic acid cycle (Denton et al., 1978; Robb-Gaspers et al., 1998), and cell death (Williams et al., 2013). In addition, the transport of Ca2+ across the inner mitochondrial membrane is an essential signaling pathway for cellular metabolic functions (Lemasters et al., 2009). However, dysregulated Ca2+ cycling excessively increases the production of mitochondrial ROS and stimulates mitochondrial Ca2+ overload, resulting in increased opening of susceptible mitochondrial permeability transition pores (mPTPs) (De Stefani et al., 2016), which preludes cell death (Panel et al., 2018). Aging reportedly induces mitochondrial Ca2+ overload, as evidenced by Ca2+ overload-induced increase in the opening of mPTPs in aged rat heart compared with young rat heart (Hunter et al., 2012).
Taken together, the data suggest that mitochondrial dysfunction could occur with aging, which would eventually induce age-related cardiac dysfunction. However, most of the previous studies regarding aging-induced alteration of mitochondrial function in cardiac muscle have been confined to isolated mitochondria, in which only mitochondrial function and structure could be impaired (Picard et al., 2011). Therefore, the purpose of this study was to determine the effects of aging on mitochondrial OXPHOS and mitochondrial function (e.g., mitochondrial H2O2 emission and Ca2+ retention capacity) in permeabilized rat heart. We hypothesized that mitochondrial OXPHOS and mitochondrial function are dysregulated by senescence in rat cardiac muscle.
MATERIALS AND METHODS
Animals
Male Fischer 344 rats were housed in a temperature- and humidity-controlled (20°C±2°C) room with a 12-hr light (8:00 a.m. to 7:59 p.m.) and dark (8:00 p.m. to 8:00 a.m.) cycle. Rats were given time to adapt to the environment for approximately 1 week in the animal cages prior to beginning the experiment. Food and water were provided ad libitum throughout the study period. The rats were divided into the VYS, YS, MS, and OS groups (n=10 per group). The experimental procedures were performed in accordance with the animal care guidelines of the National Institutes of Health and the Korean Academy of Medical Sciences.
Preparation of tissue and permeabilized muscle fiber bundles
Rats were anesthetized using ether prior to sacrifice. Hearts were extracted, weighed, and dissected to obtain the left ventricle (LV). Approximately 2 mg of LV tissue was permeabilized to assess mitochondrial functions. The other samples were snap-frozen in liquid nitrogen and stored at −80°C freezer until further analysis.
Permeabilized muscles in LV were placed in ice-cold buffer X containing 50 mM MES, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 5.7 mM ATP, 14.3 mM phosphocreatine, and 6.56 mM MgCl2-6H2O (pH, 7.1; 295 mOsm) and trimmed of fat and connective tissue. The small muscle fiber bundles were separated along the longitudinal axis and treated with 100-μg/mL saponin to increase the permeability of the sarcolemmal membrane while keeping mitochondrial membrane intact for 30 min. The permeabilized LV fiber bundles were washed in ice-cold buffer Z and added to 50 μM EGTA (washing buffer) containing 105 mM K-MES, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2-6H2O, and 0.5 mM mg/mL bovine serum albumin (pH, 7.1) for at least 15 min in a shaker and remained in buffer Z on a rotator apparatus at 4°C until analyzed. The permeabilized LV fiber bundles were used for measurements of mitochondrial H2O2 emission and Ca2+ retention capacity.
Mitochondrial H2O2 emission
Mitochondrial H2O2 emission was measured in buffer Z at 37°C (ΔF / min) during state 4 respiration (10-μg/mL oligomycin) by continuously monitoring the oxidation of Amplex Red (excitation/emission wavelengths=567/587 nm) using a SPEX Fluoromax 4 spectrofluorometer (HORIBA, Edison, NJ, USA). The procedure used 2-mg LV muscle fiber, 10 μM Amplex Red, 1.5 U/mL horseradish peroxidase (HRP), 10-μg/mL oligomycin settings, and 5 mM glutamate (complex I substrate)+2 mM malate (complex I substrate), 10 mM succinate (complex II substrate), and 10 mM glycerol-3 phosphate (lipid substrate). The H2O2 emission rate after removing the background value from each of the standard values (standard curve) was calculated from the slope of ΔF/min gradient values. After the experiment, the raw data of mitochondrial H2O2 emission were normalized using wet tissue weight. The rate of mitochondrial H2O2 emission was expressed as picomoles/min/mg wet tissue weight.
Mitochondrial Ca2+ retention capacity
Mitochondrial Ca2+ retention capacity was performed to assess the susceptibility of the mPTP opening as previously reported (Anderson et al., 2011) with modification. Briefly, after LV tissues were separated, permeabilized with saponin, and washed with washing buffer (buffer Z containing 50 μM EGTA), overlaid traces of changes in Ca2+ induced fluorescence by Calcium Green-5 N were measured continuously (ΔF/min) at 37°C during state 4 condition (10-μg/mL oligomycin) using the aforementioned Spex Fluormax 4 spectrofluorometer (HOROBA, Edison, NJ, USA). After establishing background ΔF/min (1 μM Calcium Green-5N 80 μM EGTA, 5 μM glutamate, and 2 μM malate), the reaction was initiated by the addition of Ca2+ pulses (30 μM), with excitation and emission wavelengths of 506 (excitation wavelength) and 532 (emission wavelength) nm, respectively. After completion of the experiment, the level of mitochondrial Ca2+ retention capacity was normalized by wet tissue weight. Total mitochondrial Ca2+ retention capacity was expressed as picomoles/min/mg wet tissue weight.
Western immunoblot for electron transport chain (ETC) composition in mitochondria
Frozen LV tissues were homogenized with lysis buffer containing 50 mM Tris/HCl (pH, 7.4), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2·6H2O, 1 mM EGTA, 1 mM PMSF, 1 mM Na2VO4, and 100 mM NaF in an OMNI TH homogenizer (OMNI International, NW Kennesaw, GA, USA). The homogenized tissue was centrifuged at 14,000 rpm for 30 min. Protein concentration was analyzed using a colorimetric protein assay kit (Bio-Rad, Hercules, CA, USA). Protein (20 μg) was heated at 35°C for 5 min and separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After loading for 2 hr at 110 V, the loaded proteins were electro-transferred for 1 hr at 400 mA onto a nitrocellulose membrane. After staining with Ponceau S (Sigma-Aldrich, St. Louis, MO, USA) to verify equal loading and transfer of proteins to the membranes, the membranes were blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T) for 2 hr. The membranes were incubated overnight at 4°C in primary antibody diluted 1:1,000 in TBS-T for recognition of specific target proteins of total OXPHOS rodent antibody cocktail (Abcam, Cambridge, MA, USA), which included the following: complex I (NADH dehydrogenase (ubiquinone) 1 beta subcomplex subunit 8, NDUFB8, 20 kDa), complex II (succinate dehydrogenase (ubiquinone) iron-sulfur subunit, SDHB, 30 kDa), complex III (cytochrome b-c1 complex subunit 2, UQCRC2, 48 kDa), complex IV (cytochrome c oxidase subunit 2, MTCO2, 40 kDa), and complex V (ATP synthase subunit alpha, ATP5A, 55 kDa), with β-actin (Santa Cruz Biotechnology, Dallas, TX, USA) used as the loading control. Subsequently, membranes were washed with TBS-T and incubated in a 1:3,000 dilution of HRP-conjugated secondary anti-mouse antibody (Santa Cruz Biotechnology) for 1 hr at room temperature. Following three washes in TBS-T, band detection was performed using the enhanced chemiluminescence detection kit (Thermo Fisher Scientific, Waltham, MA, USA). To compare the relative expression of proteins, the detected bands were calculated densitometrically using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA).
Statistical analyses
Statistical analyses were performed using one-way analysis of variance followed by Tukey post hoc test to assess mean differences among groups for aging effects. The results are expressed as the mean±standard error of the mean. The statistical significance level was set at P<0.05.
RESULTS
Effect of aging on body weight and heart mass
The effects of aging on body weight, heart weight, and the heart mass/body weight ratio were assessed. The results are presented in Table 1. During aging, increased body weight was found in the OS group compared with the VYS, YS, and MS groups. In addition, heart mass was significantly elevated in the YS, MS, and OS groups compared with the VYS group (P<0.05) (Table 1). Importantly, the heart mass/body weight ratio, which is the surrogate of pathophysiological aging of the heart, was significantly decreased in the OS group compared with the VYS and YS groups (P<0.05) (Table 1).
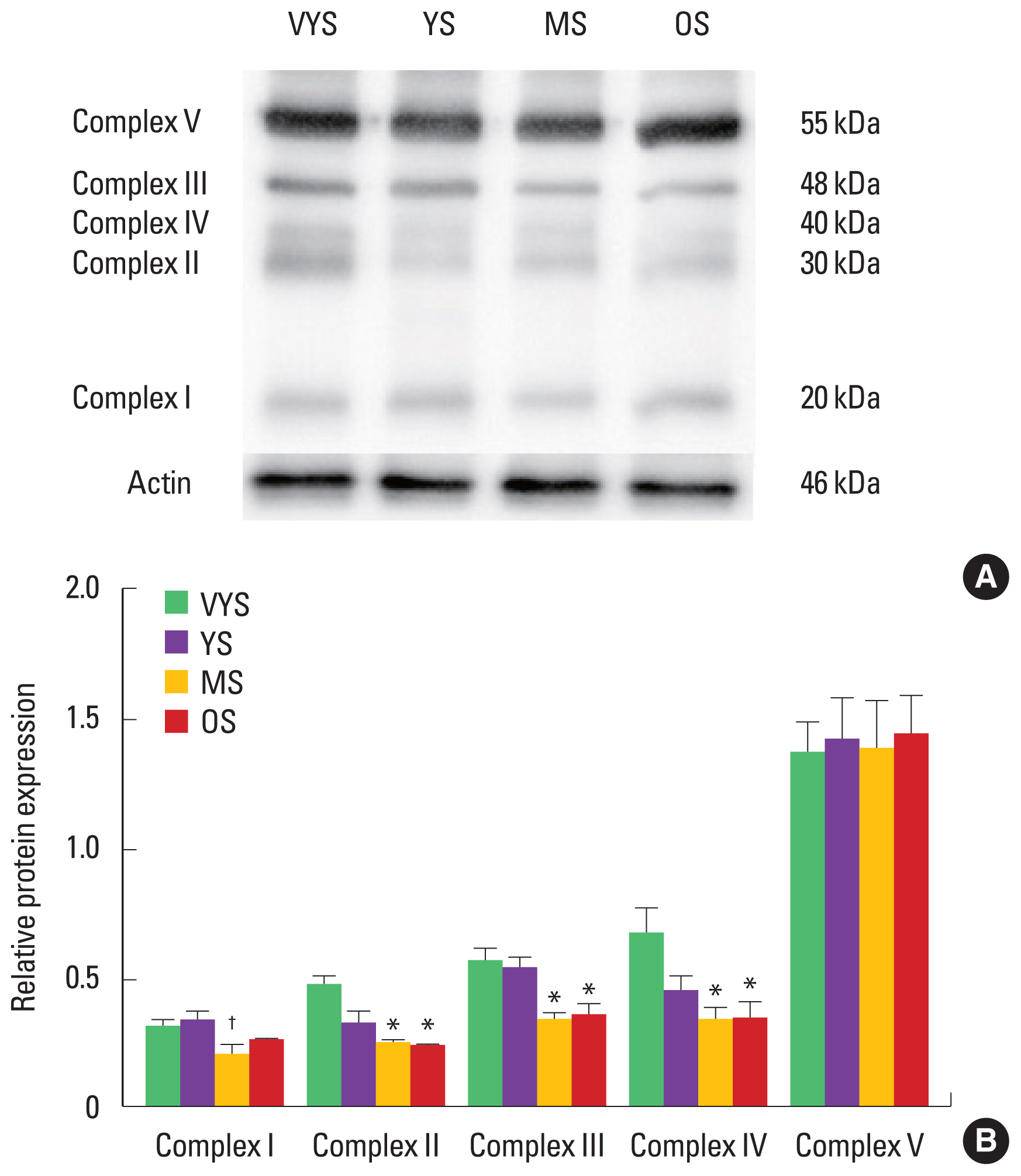
Effect of aging on protein expression of mitochondrial ETC complexes
Mitochondrial ETC complexes I–V were measured in rat heart tissues. The mitochondrial contents were decreased in the MS and OS groups compared with the VYS group, demonstrating that the protein levels of complex II (succinate dehydrogenase), III (cytochrome c reductase), and IV (cytochrome c oxidase) were significantly lowered in the MS and OS groups compared with that in the VYS group (P<0.05) (Fig. 1). Complex V (ATP synthase) was not affected by aging.
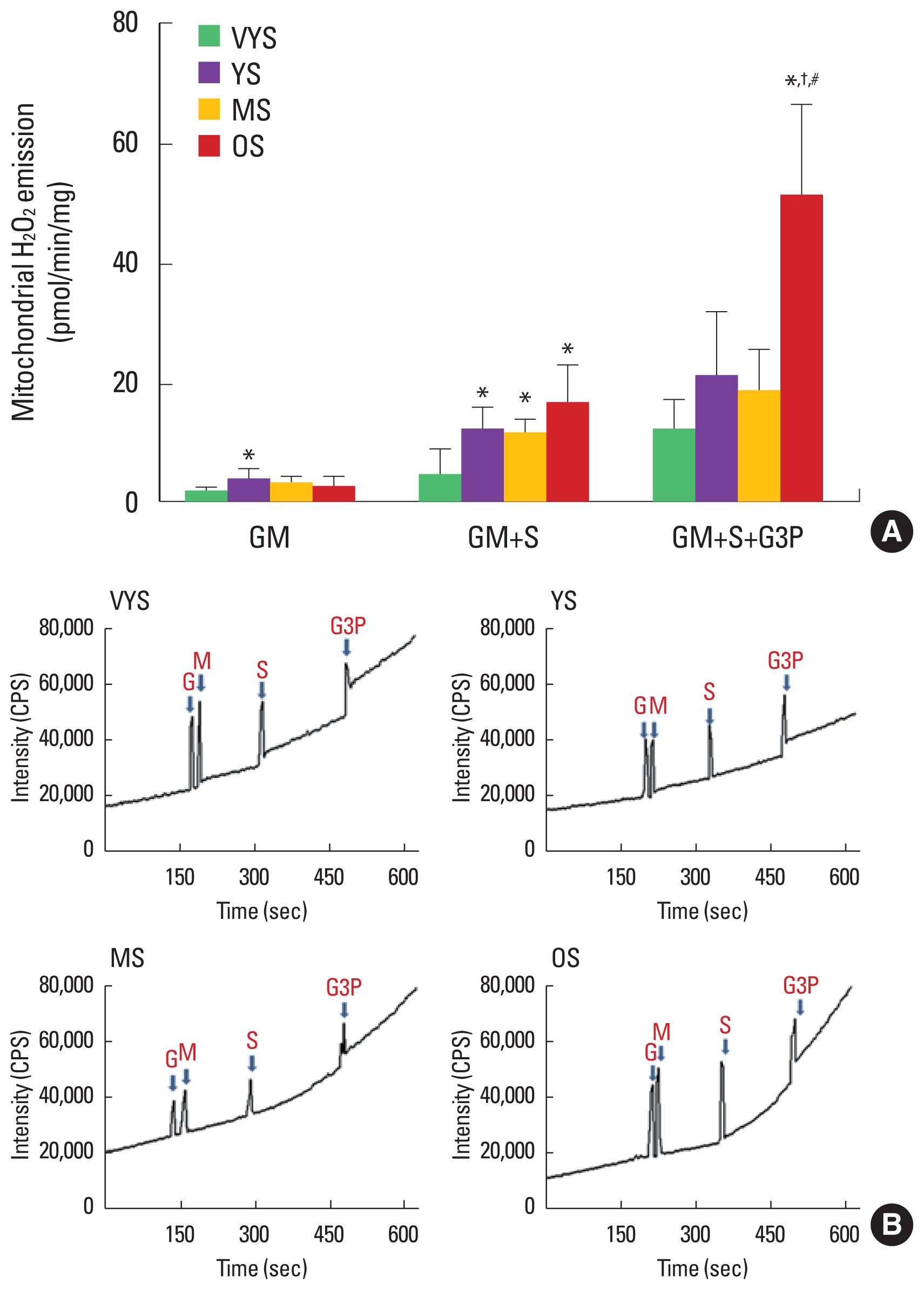
Effect of aging on mitochondrial H2O2 emission
Mitochondrial H2O2 emission was measured using glutamate+ malate (GM), succinate (S), and glycerol-3-phosphate (G3P) as substrates (Fig. 2). Mitochondrial H2O2 emission was increased only in the YS group compared with the VYS group using GM (1.05±0.32 vs. 3.38±0.66, P<0.05) (Fig. 2). In the next stage (GM+S), aging-induced mitochondrial H2O2 was observed in the OS group compared with the VYS group (4.03±1.54 vs. 16.42± 2.13, P<0.05) (Fig. 2). In addition, excessive H2O2 emission supported by GM+S+G3P was found in the OS group compared with the VYS, YS, and MS groups (11.36±1.92, 20.71±3.88, 18.11±2.63 vs. 50.82±5.55, respectively; P<0.05) (Fig. 2).
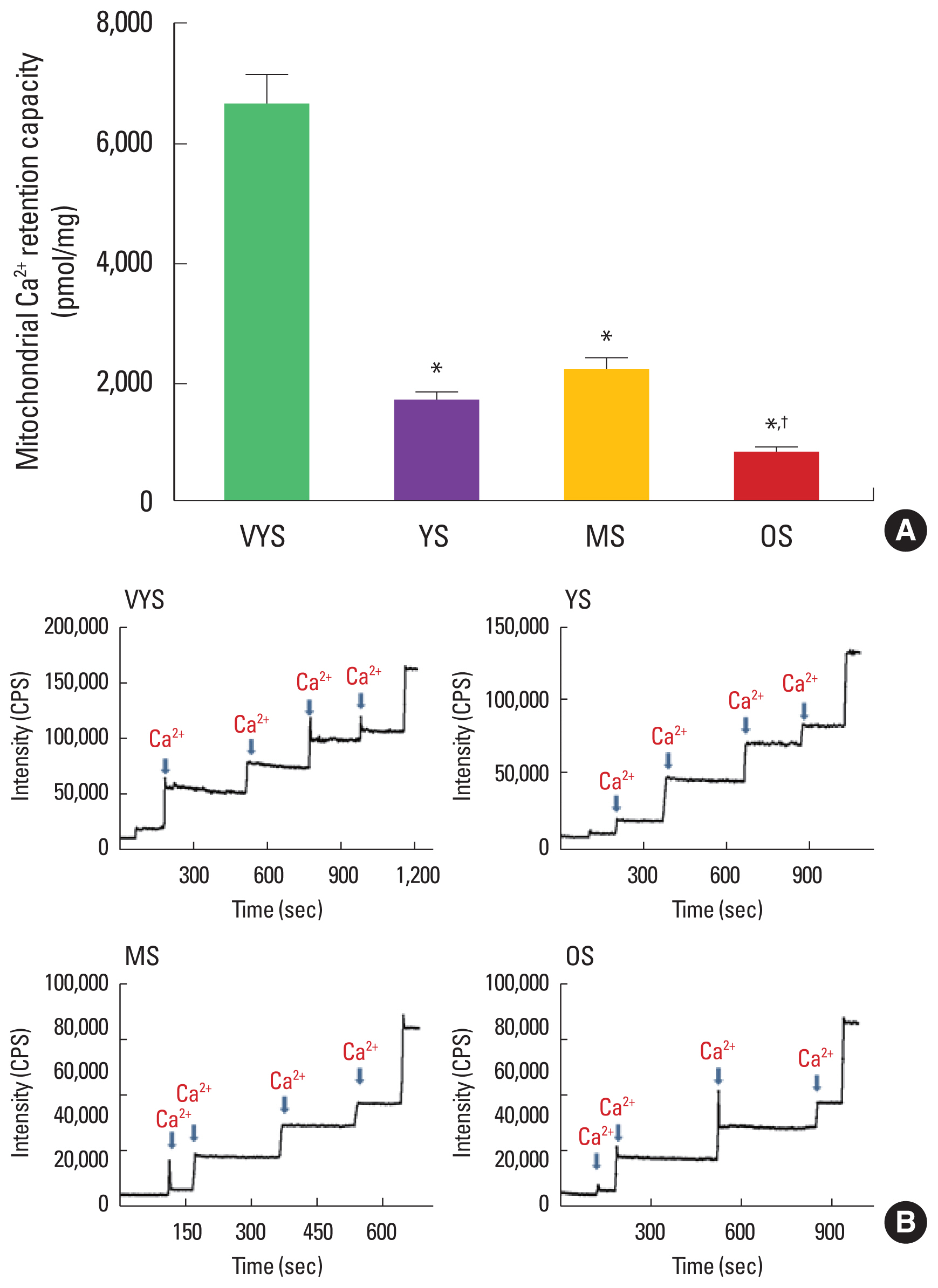
Effect of aging on mitochondrial Ca2+ retention capacity
The mitochondrial Ca2+ retention capacity was measured in permeabilized rat heart tissues. Ca2+ retention was significantly lower in the OS group compared with the VYS and MS groups (6,590.53±544.44 vs. 786.53±122.91, 2,215.14±163.46 vs. 786.53±122.91, respectively; P<0.05) (Fig. 3), suggesting that Ca2+ overload-induced mPTP opening could be activated by aging.
DISCUSSION
There are two major findings. First, aging triggered the reduced protein levels of ETC complexes in the cardiac muscles. Second, mitochondrial dysfunction (H2O2 emission and Ca2+ retention capacity) occurred as aging progressed in the permeabilized rat cardiac muscle, showing that mitochondrial H2O2 emission was excessively increased and mitochondrial Ca2+ retention capacity was progressively decreased with aging. These findings support the hypothesis that aging induces the dysregulation of mitochondrial OXPHOS and dysfunction in rat cardiac muscle.
Aging decreased the protein levels of mitochondrial ETC complexes II–IV, which are markers of mitochondrial contents, resulting in reduced energy production (Fig. 1). Age-dependent decrease in cardiac mitochondrial OXPHOS was previously associated with the decline in maximal ADP-stimulated O2 respiration (state 3) due to diminished activity of complex I and IV in mitochondrial ETC (Navarro and Boveris, 2007). In addition, reduced energy production in cardiac mitochondria contributes to increased ROS, increased mutations and deletions in the mitochondrial genome, and impaired regulation of mitochondrial biogenesis (Navarro and Boveris, 2007; Tatarková et al., 2011). Indeed, in this study, aging-induced decreases in protein levels of mitochondrial complexes were observed in complex II, III, and IV, suggesting that energy production was decreased by aging.
We also demonstrated that aging induced the mitochondria-mediated production of ROS, such as H2O2 emission in rat cardiac muscle. Previous studies have associated aging with increased mitochondrial H2O2 emission (Bejma et al., 2000; Kuka et al., 2013; Petrosillo et al., 2009). Consistent with the prior findings, we presently observed that elevated mitochondrial H2O2 emission in aging cardiac muscle compared with young cardiac muscle (Fig. 2). Specifically, the current results demonstrated that mitochondrial H2O2 emission was significantly increased in GM+S (complex II substrate) and GM+S+G3P (lipid substrate) in OS group, suggesting that this aging-induced increase in mitochondrial ROS could reuslt in the accumulation of damaged proteins, which could play a major role in generating aging-related tissue dysfunction (Balaban et al., 2005).
Finally, we also observed that mitochondrial Ca2+ retention capacity was significantly reduced by aging. Mitochondrial Ca2+ is essential in regulating ATP production by controlling OXPHOS of the heart (Kohlhaas and Maack, 2013). However, aging can exacerbate mitochondrial Ca2+ regulation, and progressively impair the myocardial Ca2+ transport system, Ca2+ storage capacities, and contractile function by altering cytosolic Ca2+ handling (Panel et al., 2018), which are associated with mitochondrial Ca2+ overload. Consistent with this recent study, we also obbserved diminshed mitochondrial Ca2+ retention capacity in aging cardiac muscle (Fig. 3), which resulted in more prevalent mPTP opening, compared with that in young cardiac muscle.
The collective data demonstrate that aging dysregulates OXPHOS, with lower protein levels of complex II–IV in the ETC in the cardiac muscle. In addition, aging is associated with induced mitochondrial dysfunction, with increased mitochondrial H2O2 emission and decreased mitochondrial Ca2+ retention capacity in permeabilized cardiac muscle fiber of aging rats. These findings demonstrate a pivotal role of aging in dysregulation of mitochondrial OXPHOS and mitochondrial function.