INTRODUCTION
Aging is characterized by a progressive decline in cardiac function including stroke volume, cardiac output, blood flow, and oxygen consumption (Beere et al., 1999; Dinenno et al., 2001; Fiechter et al., 2013) and an increase in susceptibility to inflammation, oxidative stress, and disease (Higashi et al., 2012; Sastre et al., 2000; Zhang and Herman, 2002). Impaired cardiac function, increased inflammatory cytokines, and elevated oxidative stress are commonalities between aging and cardiovascular disease. A significant reduction in the number of myocytes is now believed to contribute directly to impaired contractile function, cardiomyopathy, heart disease, and heart failure (Favaloro et al., 2012; Kajstura et al., 1996; Pollack et al., 2002). Indeed, the typical 70 year old man has a 30% reduction in the number of cardiac myocytes compared with young adults (Higami and Shimokawa, 2000; Olivetti et al., 1991).
Loss of myocytes with aging occurs through necrosis (i.e., accidental cell death) and apoptosis (i.e., programmed cell death), two distinct mechanisms leading to cell death (Konstantinidis et al., 2012). Necrosis results from cellular injury seen with infection or inflammatory disease, and is characterized by cellular swelling and rupturing with inflammation (Carraro and Franceschi, 1997; Mughal et al., 2012; Phaneuf and Leeuwenburgh, 2001; Zhang and Herman, 2002). Cell rupture with necrosis results in the release of stress and inflammatory proteins and substrates. In contrast, apoptosis is highly regulated cell death without injury and inflammation, and is characterized by cell shrinking/blebbing and condensation of the nucleus (Carraro and Franceschi, 1997; Mughal et al., 2012; Pollack and Leeuwenburgh, 2001; Zhang and Herman, 2002). Apoptosis is a highly regulated, programmed means of cell death or elimination that plays an essential role in governing development, growth, and repair (Hengartner, 2000; Jacobson et al., 1997; Zhang and Herman, 2002). However, excessive apoptosis results in dysfunction and disease. A dramatic increase in the rate of apoptosis has been reported with aging in the rat left ventricle, while the rate of necrosis remained constant (Higami and Shimokawa, 2000; Kajstura et al., 1996; Konstantinidis et al., 2012; Liu et al., 2012). Phaneuf and Leeuwenburgh (2002) described age-related disruption of Bcl-2 family signaling in the rat heart. Progressive apoptosis from age in post-mitotic tissues such as the heart is dire, as lost myocytes are not replaced. Unfortunately, the mechanisms responsible for apoptotic signaling and apoptosis in the aging heart remain unclear and very limited.
In contrast, exercise training can reduce the risk of injury, oxidative stress, and inflammatory signaling from mechanical and oxidant perturbations, as long as overtraining does not occur (Fielding and Evans, 1997; Hammeren et al., 1992; Papathanasiou et al., 2012; Powers et al., 2002). Also, exercise training improves cardiovascular capacity and reduces cardiovascular disease risk in both young adults and the elderly (Booth et al., 2002; Gremeaux et al., 2012; Lawler et al., 1993). Exercise has the potential to reduce apoptosis through upregulation of protective stress-sensitive proteins including nuclear factor kappaB (NF-kB), insulin-like growth factor (IGF-1), and heat shock proteins (HSP90 and HSP70) (Frost and Lang, 2003; Milne and Noble, 2002; Morton et al, 2009; Naito et al., 2001). However, the mechanisms by which exercise training improves heart function and cardiovascular disease risk profile are not well delineated. Moreover, the ability of exercise training to modulate Bcl-2 family apoptotic signaling and apoptosis in the aging heart has not been clearly explored so far. Therefore, in the next section, an overview of aging and apoptosis in the heart will be provided, followed by a brief review of the literature supporting the effects of exercise in the aging heart.
APOPTOSIS AND MITOCHONDRIAL CONTROL
Apoptosis is regulated by genetic programming and plays an essential role in governing development, growth, and disease (Hengartner, 2000; Zhang and Herman, 2002). One of the most visible examples of cell death that occurs in normal animal development is the loss of the tadpole’s tail (Jacobson et al., 1997). However, excessive apoptosis or dysregulation of apoptosis results in dysfunction and disease (Zhang and Herman, 2002).
Apoptosis is distinct from necrosis (Carraro and Franceschi, 1997; Fiers et al., 1998; Konstantinidis et al., 2012; Pollack and Leewenburgh, 2001). Necrosis or “accidental cell death” is the pathological process which is caused by injury, infection or inflammatory diseases such as high blood pressure and heart attacks (Hofmann, 1999). In contrast, apoptosis or “programmed cell death” is highly regulated cell death involving DNA fragmentation, blebbing, and dismantling of the cell without inflammation (Favaloro et al., 2012; Jacobson et al., 1997; Lemasters et al., 1999).
Apoptotic signaling induces apoptosis through caspase-dependent pathways (Budihardjo et al., 1999; Favaloro et al., 2012; Hengartner, 2000) (Fig. 1). They include (a) cytokines/Fas-driven pathways (Hofmann, 1999), (b) mitochondrial-driven pathways (Mignotte and Vayssiere, 1998), and (c) endoplasmic reticulum (ER)/Ca2+-driven pathways (Pollack and Leeuwenburgh, 2001). Binding of cytokines (e.g., TNF-α, IL-1β) or Fas to their cell membrane receptors initiate apoptotic pathway through the recruitment of adaptor proteins (e.g., Fas-associated death domain; FADD). The resulting death-inducing signaling complex activates caspase-8, which subsequently activates caspase-3 (Hofmann, 1999). Endoplasmic reticulum (ER)-mediated apoptotic signaling pathway is initiated by the involvement of capase-12, which also subsequently activates caspase-3 (Pollack and Leeuwenburgh, 2001). Mitochondrial-mediated pathways including the Bcl-2 family is the best characterized, and Bcl-2 signaling is believed critical in regulating apoptosis with aging (Green and Reed, 1998; Hengartner, 2000; Konstantinidis et al., 2012; Mignotte and Vayssiere, 1998; Pollack and Leeuwenburgh, 2001). In other words, mitochondria are important sites of programmed cell death (Favaloro et al., 2012; Remmen and Richardson, 2001). A critical factor in mitochondrial-mediated apoptosis is that deficiency of survival stimuli such as growth factors increases mitochondrial permeability. Mitochondrial membrane permeability transition pore opening is regulated by a number of pro-and anti-apoptotic proteins such as Bcl-2 family of proteins (Green and Reed, 1998; Hengartner, 2000; Mignotte and Vayssiere, 1998).
Bcl-2 family has been classified into three functional groups (Hengartner, 2000). Group I such as Bcl-2, Bcl-XL inhibits apoptosis. Group II such as Bax initiates apoptosis. And group III such as Bid also initiates apoptosis. In other words, Group I functions as gatekeepers at the outer surface of mitochondria, whereas Group II and III function as gatecrashers. Therefore, cell death or survival depends on the ratio of the pro- and anti-apoptotic proteins expressed. For example, high levels of Bcl-2 relative to Bax promote survival, whereas the reverse ratio promotes myonuclear cell death (Hengartner, 2000; Pawlowski and Kraft, 2000; Primeau et al., 2002). Thus the ratio of anti-apoptotic/pro-apoptotic proteins in the Bcl-2 family functions as an upstream modulator of apoptosis in the mitochondrial-mediated apoptotic pathways. The proteins of Bcl-2 family can regulate apoptosis by controlling permeability transition pore (PTP) opening and the release of cytochrome c from mitochondria (Mignotte and Vayssiere, 1998).
Caspases are a group of cysteine dependent aspartate-specific proteases (Favaloro et al., 2012; Nicholson, 1999; Zhang et al., 2003). In other words, caspases are a complex cascade of protein- cleaving enzymes. Fourteen caspases have now been identified in mammals, leading to apoptotic cell death (Kumar, 1999). Caspases play a pivotal role in apoptotic pathways and interact with the non-caspase apoptotic pathways including endonuclease G (Endo G) and apoptosis inducing factor (AIF) (Marzetti et al., 2012; Nicholson, 1999; Zhang et al., 2003). Cytochrome c associates with Apaf-1 (apoptotic protease-activating factor 1) and pro-cas pase-9 to form the apoptosome, which activates caspase-9 and caspase-3, resulting in apoptosis through DNA fragmentation (Favaloro et al., 2012; Kumar et al., 2002; Mignotte and Vayssiere, 1998; Pollack et al., 2002; Susin et al., 2000; Zhang et al., 2002).
AGING AND APOPTOSIS
Aging is characterized by a general decline of physiological function (Dinenno et al., 2001; Liu et al., 2012). Aging enhances the susceptibility to apoptosis in several types of tissues. It has been shown that aging is associated with increased Bax protein as well as enhanced DNA fragmentation in brains (Higami and Shimokawa, 2000). Aging-induced apoptosis may contribute to a 30% loss of cardiac myocytes outside changes noted in cardiac diseases (Higami and Shimokawa, 2000; Liu et al., 2012; Olivetti et al., 1991). Myocyte cell death including both apoptosis and necrosis increased with aging in the heart of Fischer-344 rats, mediating ventricular dysfunction and failure (Kajstura et al., 1996). Liu et al. (1998) also reported that expression of Bcl-2 and Bax proteins both increased in the heart of elderly Fischer-344 rats under physiological conditions. Moreover, our research group demonstrated that aging increased pro-apoptotic Bax, cleaved caspase-3, and DNA fragmentation in rat skeletal muscle (Song et al. 2006) as well as heart (Kwak et al., 2006). In contrast, Nitahara et al. (1998) showed that there was an increase in cardiomyocyte apoptosis with age from Fischer-344 rats without significant changes in Bax and Bcl-2 protein levels with age. These findings suggest that age-related changes in the expression of Bcl-2 family proteins (Bcl-2 and Bax) are still debatable.
Progressive apoptosis with advancing age in post-mitotic tissues including heart, skeletal muscle, and brain is dire, because lost myocytes are not replaced. Phaneuf and Leeuwenburgh (2002) showed markers of apoptosis in hearts (left and right ventricles) of 6-, 16-, and 24-month-old male Fischer-344 rats. Cytosolic cyto-chrome c was significantly increased in the 16- and 24-month-old animals compared to the 6-month-old animals, indicating increased mitochondrial permeability transition pore (PTP) opening and activation of the apoptosome. Also, Bcl-2 levels were decreased with aging, whereas mitochondrial Bax levels remained unchanged.
In contrast, mitotic tissues such as the liver, colon mucosa, etc. display reduced pro-apoptotic protein levels, cytochrome c contents, caspase levels, and DNA fragmentation with aging. This dysregulation of apoptotic signaling from aging is postulated as a mechanism contributing to cancer risk (Suh et al., 2002). For example, it has been reported that caspase-3 and caspase-9 protein levels and activities were lower in colon mucosa in old (22–24 months) Fischer-344 rats compared to their young (4–5 months) and middle-age (12–14 months) counterparts (Xiao et al., 2001). These changes were accompanied by a reduced number of apoptotic cells in the colonic mucosa of 12–14 and 22–24 month old rats compared with in 4–6 month old rats. In addition, the levels of pro-apoptotic Bak were decreased by about 50%, whereas anti-apoptotic Bcl-XL levels were increased by about 50% in 22 month old rats compared to 4 or 13 month old rats (Xiao et al., 2001). Suh et al. (2002) also showed that apoptosis-induced DNA fragmentation was dramatically upregulated in the liver of young (2 months) female Fischer rats but not in the old (26 months) counterparts. Additionally, the levels of cytochrome c in livers from old (26 months) Fischer-344 rats were significantly lower than those from young (6 months) counterparts (Zhang et al., 2002). These opposite results could be tissue specific and possible characteristics of mitotic but not post-mitotic tissues.
APOPTOSIS AND HEART
The heart may undergo an increase in apoptosis during ischemia-reperfusion, heart failure, doxorubicin treatment, and aging (Childs et al., 2002; Higami and Shimokawa, 2000; Olivetti et al., 1997; Powers et al., 2002; Tacar et al., 2013). It has been reported that a number of cardiomyopathies are associated with mitochondrial DNA damage which leads to defects in electron transport chain and the upregulation of ROS (Anderson et al, 2011; Mignotte and Vayssiere, 1998), resulting in the apoptosis of cardiac myocytes (Li et al., 2012; Mughal et al., 2012; Von Harsdorf et al., 1999). Heart is an energetically-demanding tissue that requires a continuous production of ATP via mitochondrial respiration (Li et al., 2012; Primeau et al., 2002). In highly aerobic tissues such as heart, mitochondria occupy up to 25% of cell volume (Lemasters et al., 1999; Li et al., 2012). It was also shown that tumor necrosis factor-α (TNF-α) increased apoptosis via inducible nitric oxide synthase (iNOS) expression and nitric oxide (NO) in cardiac myocytes (Ing et al., 1999; Song et al., 2000). In both humans and animals, the aging heart is characterized by a decrease in the total number of myocytes resulting in reactive hypertrophy of the remaining cells and increased fibrosis (Kwak et al., 2006, 2011; Olivetti et al., 1991; Phaneuf and Leeuwenburgh, 2002).
Ischemia and reperfusion of myocardium induces apoptotic cell death (Powers et al., 2002), with direct relevance to heart disease and heart attacks (Quindry et al., 2012). Interestingly, brief periods of acute myocardial ischemia, or “preconditioning,” protects the heart from the deleterious effects of longer periods of ischemia and reperfusion (Murry et al., 1986; Taylor and Starnes, 2003). The mechanisms of protection against apoptosis due to preconditioning may be mediated by NF-kB, which inhibits the susceptibility to apoptosis and can promote upregulation of anti-apoptotic Bcl-2 (Das et al., 1999; Maulik et al., 1998). Indeed, NF-kB DNA binding activity progressively increased as a function of the pre- conditioning of ischemia (Maulik et al., 2000), and Bcl-2 levels were decreased in the ischemic/reperfused heart, but increased in the preconditioned myocardium (Maulik et al., 2000). In addition, overexpression of anti-apoptotic Bcl-2 was effective in reducing myocardial reperfusion injury and improving heart function (Brocheriou et al., 2000).
Indeed, the balance between Bax/Bcl-2 is postulated as a critically important factor in the increased rate of apoptosis in cardiac myocytes (Condorelli et al., 1999; Pollack et al., 2002). For example, Condorelli et al. (1999) indicated that left ventricular hypertrophy and left ventricular dysfunction in a rat model of chronic pressure overload were accompanied by upregulated pro-apoptotic Bax and Bax/Bcl-2 ratio, leading to cardiomyocyte apoptosis. Kang et al. (2000) also found that apoptosis predominated in cardiomyocytes after reoxygenation through a mitochondrial apoptotic pathway, and that Bcl-2 prevented reoxygenation-induced apoptosis by inhibiting the release of cytochrome c from mitochondria. In addition, Kirshenbaum and Moissac (1997) concluded that the anti-apoptotic Bcl-2 protein can prevent apoptosis of ventricular myocytes.
Growing evidence shows that cardiomyocyte apoptosis also contributes to congestive heart failure. For example, Olivetti et al. (1997) showed that cardiomyocyte apoptosis dramatically increased in the heart of patients with cardiac failure, although the level of Bcl-2 was about two times higher than that of the normal hearts, while the level of Bax remained unchanged. Saraste el al. (1999) also showed that the number of cardiomyocytes undergoing apoptosis was significantly increased in failing human hearts compared with control myocardium, and that the expression of Bcl-2 was increased in failing hearts. Enhanced expression of Bcl-2 in the failing heart suggested that compensatory mechanisms were activated to prevent apoptosis (Saraste et al., 1999). The expression of Bcl-2 protein was induced in human cardiac myocytes at the acute stage of infarction, but the expression of Bax protein was overexpressed at the old stage, which was related to myocyte death in human hearts (Misao et al., 1996).
EXERCISE AND APOPTOSIS IN THE AGING HEART
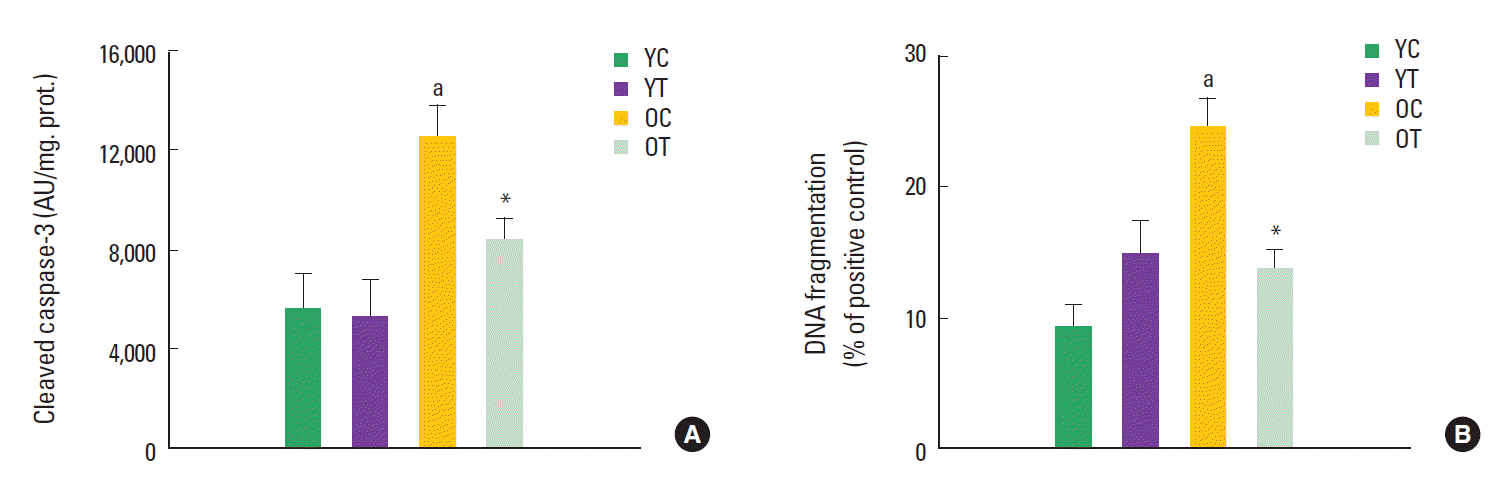
Our research group recently indicated that the Bax/Bcl-2 ratio was significantly upregulated with age and markedly reduced by exercise training in the left ventricles (Kwak et al., 2006). These data imply that aging heart is more susceptible to apoptosis than young heart, and that chronic exercise training exerts anti-apoptotic action in the aging heart (Soufi et al., 2008). These data are consistent with previous reports showing that aging is associated with increased Bax protein as well as decreased Bcl-2 (Higami and Shimokawa, 2000; Liu et al., 1998), indicating that mitochondrial Bcl-2 family signaling is an important site of regulating apoptosis with aging and exercise training.
Based on our data (Kwak et al., 2006), the increased Bax/Bcl-2 ratio with aging heart was directly related to increases in downstream caspase signaling including caspase-9 and cleaved caspase-3, which promote DNA fragmentation and cell death (e.g., increased TUNEL-positive nuclei). Similarly, there were consistent reports that caspase-9 and caspase-3 activities were increased in aging rat liver (Zhang and Herman, 2002). In contrast, exercise training markedly decreased caspase-9 and cleaved caspase-3 levels as well as the Bax/Bcl-2 ratio in the aging heart, which presumably led to the decreased DNA fragmentation. These results are consistent with the hypotheses that aging increases Bcl-2 family pro-apoptotic signaling in the heart, and exercise training in the aging heart results in amelioration of age-induced changes in the mitochondrial-mediated apoptotic pathways. In particular, cleaved caspase-3 showed similar patterns to DNA fragmentation in the effect of aging and exercise training, suggesting that caspase-3 may be an essential hallmark of apoptosis (Fig. 2).
Aging is generally characterized by a decline of cardiac function and an upregulation of oxidative stress in the heart (Beere et al., 1999; Dinenn et al., 2001; Sastre et al., 2000; Soufi et al., 2008), which are major contributors to cell death through mitochondrial dysfunction (Dirks and Leeuwenburgh, 2002; Sastre et al., 2000). Previous work has demonstrated that aging promotes the susceptibility to apoptosis in rat heart (Higami and Shimokawa, 2000; Kajstura et al., 1996; Nitahara et al. 1998; Olivetti et al., 1991; Soufi et al., 2008). While aging heart was vulnerable to apoptosis, exercise training was effective in diminishing apoptosis (DNA fragmentation) in the aging heart, which indicated the beneficial effect of long-term exercise training on apoptosis in the aging heart (Soufi et al., 2008). Interestingly we found that even if apoptosis was dramatically increased with aging, there was no significant changes in heart-to-body weight ratio between young sedentary rats and old sedentary rats (Kwak et al., 2006) because the aging process of the heart in humans and animals is characterized by a significant loss of myocytes and reactive hypertrophy of the remaining cells (Olivetti et al., 1991; Phaneuf and Leeuwenburgh, 2002). Thus, the loss of cardiac myoctyes doesn’t necessarily cause a reduction in mass. In addition, with aging there is an increase in heart connective tissue, increasing wall stiffness/thickness, and losing elasticity of the heart (Anversa et al., 1990; Kwak et al., 2011; Lakatta and Sollott, 2002). However, cardiac myocytes that hypertrophy often have poor contractile function due to increased left ventricle wall thickness (Lakatta and Sollott, 2002).
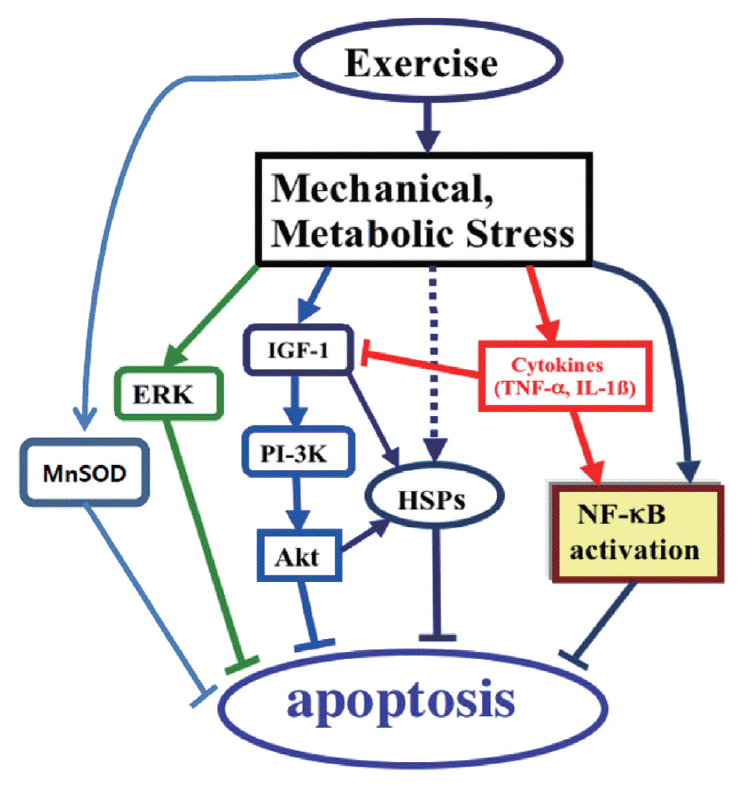
Based upon current findings regarding exercise and apoptosis in the aging heart, further investigations as future directions are required to determine the effects of exercise training on the upstream cell protective mechanisms in the aging heart (Fig. 3). For example, exercise training may promote cell-survival proteins including Mn isoform of superoxide dismutase (MnSOD), NF-kB, extracellular receptor kinase (ERK), IGF-1/Akt pathway, and heat shock proteins (HSPs) in heart (Higashi et al., 2012; Powers et al., 2002; Soufi et al., 2008; Starnes et al., 2003; Taylor and Starnes, 2003), which may be potential upstream regulators of age-induced apoptosis in the aging heart.
CONCLUSIONS
In summary, aging resulted in increases in mitochondrial-mediated apoptotic pathways including pro-apoptotic protein levels such as Bax, Bax/Bcl-2 ratio, caspase-9, and cleaved caspase-3 and apoptosis. However, endurance exercise training reversed the elevation of apoptotic signaling and apoptosis, suggesting that exercise training protects the heart against apoptosis. However, further research is necessary to determine the cellular and molecular mechanisms by which exercise training protects against aging-induced apoptosis in the heart.