Regulation of blood flow in small arteries: mechanosensory events underlying myogenic vasoconstriction
Article information
Abstract
As blood flow is proportional to the fourth power of the vascular radius small changes in the diameter of resistance arteries/arterioles following an increase in intraluminal pressure would be expected to substantially increase blood flow. However, arteriolar myocytes display an intrinsic ability to locally regulate blood flow according to metabolic demands by tuning the diameter of small arteries in response to local changes in he-modynamics. Critical to this, observations were made more than 100 years ago that mechanosensitive small arteries exhibit the “myogenic response” or pressure-induced vasoconstriction or vasodilation in re-sponse to increased or decreased intravascular pressure, respectively. Although cellular mechanisms underlying the myogenic response have now been studied extensively, the precise cellular mechanisms under-lying this intriguing phenomenon still remain uncertain. In particular, the biological machinery that senses changes in intravascular pressure in vascular smooth muscle cells have not been unquestionably identified and remain a significant issue in vascular biology to be fully elucidated. As such, this brief review focuses on putative mechanosensors that have been proposed to contribute to myogenic vasoreactivity. Specific attention is paid to the roles of integrins, G protein-coupled receptors, and cadherins.
INTRODUCTION
Functionally active tissues typically exhibit increased metabolic demands (e.g. an accelerated glucose disappearance, high oxygen consumption) that require marked increases in blood flow. This is most evident in active skeletal muscle where the functional response is termed exercise hyperemia where blood flow can increase many folds over basal levels. Conversely, blood flow in quiescent skeletal muscle is typically relatively lower compared with highly metabolic organs including the heart, kidney, and brain. Blood flow to (non)contracting skeletal muscle is generally determined by vascular resistance. The low blood flow under resting conditions in skeletal muscle is attributed to relatively high vascular resistance/tone that results from an inherent ability of resistance arteries and arterioles to respond to changes in intravascular pressure along with sympathetic nerve activation. Specifically, vascular smooth muscle cells (VSMC) constrict when intravascular pressure increases, called myogenic vasoconstriction or pressure-induced vasoconstriction. The myogenically-active arteries and arterioles in quiescent skeletal muscle play a significant role in regulating local blood flow according to metabolic demands and maintaining appropriate blood distribution and perfusion pressure to many other organs (Clifford, 2011). Thus, an appreciation of the cellular mechanisms underlying these vasomotor responses is critical to understanding the basic functions of skeletal muscle. Further, disorders of local vascular control likely contribute to vascular pathophysiology including exercise intolerance.
While pressure-induced, myogenic, vasoconstriction in skeletal muscle resistance vessels is known to be dependent on VSMC depolarization, increased Ca2+ entry via voltage-operated calcium channels and subsequent phosphorylation of the 20 kDa myosin regulatory light chain (Fig. 1), the events or biological sensors involved in the upstream ‘mechanosensation’ are less clearly understood. Substantial interest has been devoted to the involvement of mechanosensitive ion channels as an underlying mechanism for initiation of depolarization (Bulley et al., 2012; Davis et al., 1992; Drummond et al., 2004; Harder, 1984; Jaggar et al., 1998; Kotecha and Hill, 2005; Welsh et al., 2002; Wu and Davis, 2001). More recently, evidence has mounted for an additional step, which suggests that classes of G protein-coupled receptor (GPCR) can be activated directly by mechanical stimuli – thus not requiring their classical ligands to initiate signaling events. A further consequence of mechanically induced GPCR activation, yet to be experimentally considered, is that receptor-transactivation leads to the stimulation of pathways that regulate a number of cellular events in parallel to contraction (detailed in Fig. 1 but not substantially addressed in this brief presentation). Moreover, proteins in the extracellular matrix environment or intercellular junctions are now drawing attention as potential biological mechanosensors transmitting physical stress for conversion to biological events (i.e., the myogenic response) (Hill and Meininger, 2012). Thus, putative biological sensors within arteriolar myocytes are described in this review.

Schematic diagram outlining signaling events underlying pressure-induced vasoconstriction. Mechanical stimuli on arteriolar wall generated by an increase in intraluminal pressure induce VSMC deformation (e.g., longitudinally-stretched VSMC membrane), which is detected by mechanosensitive extracellular matrix proteins, transmembrane ion channels, and receptors. The detected mechanical stimuli are transduced into biological signaling events underlying the arteriolar myogenic reactivity. Although emphasis in this paper is placed on the acute mechanosensory events, it is apparent that mechanoactivation involves a number of temporally specific events including longer-term adaptive events that may be evident as vascular remodeling. Collectively, this plasticity contributes to local blood flow control over acute and more chronic time frames. BKCa, large conductance Ca2+-activated K+ channels; ENaC, epithelial sodium channels; Kv, voltage-dependent K+ channels; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; PKC, protein kinase C; RhoK, Rho kinases; SAC, stretch-activated ion channels; TRP, transient receptor potential ion channels; VSMC, vascular smooth muscle cell.
ROLES OF INTEGRINS AND CADHERINS IN MYOGENIC CONSTRICTION
Integrins appear as a class of strong candidate mechanosensory proteins as they provide a link between the extracellular matrix, plasma membrane, and the myocyte cytoskeleton and ultimately the contractile machinery (Fig. 2A). In effect, they are a cell-surface-adhesion receptor comprising a noncovalent association of α- and β-subunit heterodimers. Among a variety of combinations for the heterodimers (more than 24 distinct integrins being described), the predominant species reported in arteriolar myocytes are α1β1-, α3β1-, α4β1-, α5β1-, αvβ1-, αvβ3-, and αvβ5 (Davis et al., 2001; Martinez-Lemus et al., 2005). In regard to vasomotor responses, integrin-recognizing synthetic peptides containing the tri-peptide arginine-glycine-aspartate (RGD) induce a sustained vasodilation in rat cremaster arterioles (Mogford et al., 1996) with the RGD peptide-mediated dilation being associated with a decrease in intracellular Ca2+ (D’Angelo et al., 1997; Pierschbacher and Ruoslahti, 1987). However, the impact of RGD peptide on vascular reactivity is complex as the peptide causes vasoconstriction of rat renal afferent arterioles that appears to be mediated via an increase in intracellular Ca2+ (Yip and Marsh, 1997). Given this discrepancy studies have been undertaken to delineate the effect of integrin binding on key smooth muscle ion channels. Soluble ligands of α5β1 integrin (i.e., RGD, fibronectin) are thus shown to lead to increased Ca2+ influx via dihydropyridine-sensitive Ca2+ channels, whereas ligand-induced activation of αvβ3 integrin reduces Ca2+ entry (Wu et al., 1998). The reciprocal Ca2+ regulation of these integrins may be related to divergent endpoint responses such as vasoconstriction or dilation. In addition to integrin-mediated direct regulation of Ca2+ influx, integrins have an ability to indirectly modulate Ca2+ signaling, vasomotor reactivity, or even myogenic responsiveness by enhancing K+ efflux through large conductance Ca2+-activated K+ (BKCa) channels. Consistent with this, it has been shown that activation of α5β1 integrin facilitates BKCa activation via c-src-dependent phosphorylation of BKCa (Wu et al., 2008; Yang et al., 2010). Thus, it is implied that the integrin/K+ channel-induced hyperpolarization may result in inhibition of voltage-operated calcium channels and in turn alter vasomotor reactivity and/or myogenic responsiveness.
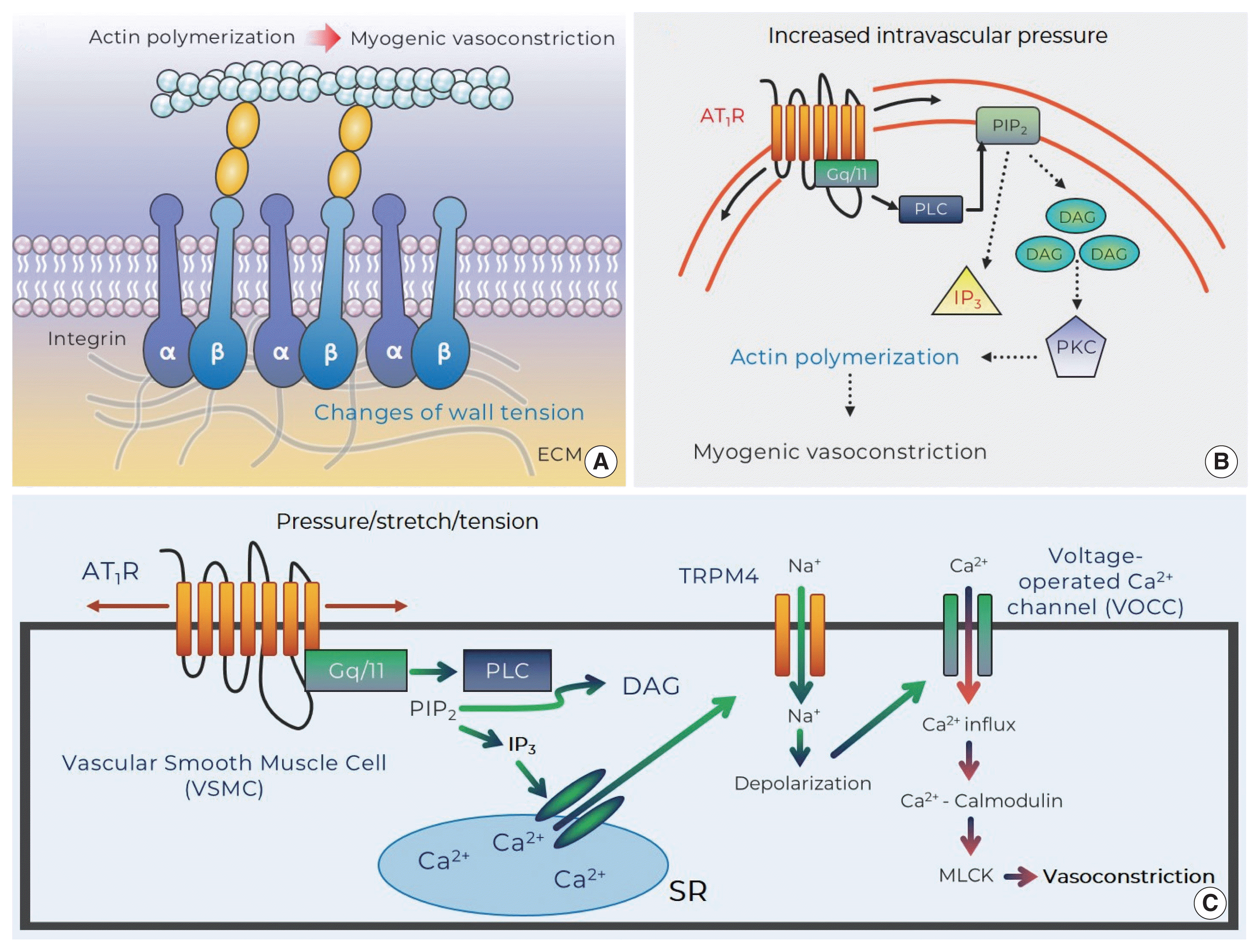
Potential mechanosensors that contribute to myogenic responsiveness. (A) Changes of vascular wall tension activate integrins and evoke myogenic vasoconstriction through cytoskeleton remodeling (i.e., actin polymerization: monomeric G-actin is incorporated into F-actins). (B) Mechanosensitive AT1R that senses increased intraluminal pressure generates DAG via PLC-dependent cleavage of PIP2. DAG stimulates PKC that induces actin cytoskeleton reorganization for myogenic vasoconstriction. (C) Stretch of VSMC provokes AT1R-mediated IP3 production and subsequently results in Ca2+ release from SR. This signaling then induces TRPM4-mediated depolarization (via Na+ entry) that contributes to Ca2+ influx-calmodulin-MLCK-dependent vasoconstriction. AT1R, angiotensin II type 1 receptor; DAG, diacylglycerol; ECM, extracellular matrix; IP3, inositol trisphosphate; MLCK, myosin light chain kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PLC, phospholipase C; SR, sarcoplasmic reticulum; TRPM4, transient receptor potential melastatin member 4.
As integrins not only interact with extracellular matrix proteins including fibronectin, collagen, vitronectin, elastin, or laminin but also intracellularly provide a platform for focal adhesion proteins (Davis et al., 2001; Vuori, 1998), they serve as direct linkers transmitting biological signals bidirectionally between extra- and intracellular environment (i.e., outside-in and inside-out signaling) (Hill and Meininger, 2012). For example, mechanical forces (e.g., stretch or tension) exerted by increased intravascular pressure are transmitted through integrins and in turn converted to intracellular signals. An intriguing question related to the role of integrins in VSMC mechanotransduction is, do integrins participate in the myogenic response? Antibodies-blocking αvβ3 or α5β1 integrin significantly abrogate pressure-induced vasoconstriction of rat cremaster arterioles (Martinez-Lemus et al., 2005). In addition, it has been observed that a genetic depletion of β1 integrin subunit profoundly reduces vascular contractile responses to norepinephrine and high concentration extracellular K+ buffer (80 mmol/L) in isolated superior mesenteric and femoral arteries as well as cremaster arterioles (1A–3A branches) in vivo (Turlo et al., 2013). More recently, it has been found that blockade of αvβ3 significantly decreases Ca2+ waves and pressure-induced vasoconstriction in cerebral arteries (Mufti et al., 2015). Therefore, these intriguing studies appear to support mechanosensitivity of integrins following an increase in intravascular pressure and thus their significant contribution to myogenic constriction.
In more recent studies, direct evidence has been sought for mechanisms by which pressure-induced stretch of arteriolar myocytes results in integrin adhesion and what are the underlying downstream signaling events, including tyrosine phosphorylation of the focal adhesion proteins, which mediate myogenic constriction. Such studies have been facilitated by the development of techniques such as atomic force microscopy which enables protein-protein interactions to be studied. In single arteriolar myocyte studies, contact with fibronectin (extracellular matrix protein)-coated atomic force microscopy probes results in the clustering of α5 and β3 integrins, consistent with focal adhesions being formed at the cytoplasmic tails of the integrins (Sun et al., 2008). Local membrane stretch of single arteriolar myocytes, induced by controlled retraction of the atomic force microscopy probes, intriguingly gives rise to myogenic-like behaviors (i.e., a counteracting pulling-down force) that are abolished by cytochalastin D (an agent for actin depolymerization) or blockade of α5β1- and αvβ3-integrins (Sun et al., 2008). Moreover, more recent investigations, using high-sensitive Western blotting techniques, have observed that pressure-dependent stretch of cerebral arteriolar myocytes (in response to a step increase in intraluminal pressure protocol) leads to α5 integrin-mediated phosphorylation of kinase proteins within integrin adhesion complexes including focal adhesion kinase and Src family kinase (Colinas et al., 2015). The phosphorylation subsequently is thought to activate adhesion scaffolding (e.g., vinculin, paxillin) and signaling (e.g., phospholipase C gamma1) proteins. In parallel with these mechanisms, stimulation of protein kinase C and Rho-associated kinase gives rise to myosin phosphatase target subunit 1-mediated Ca2+ sensitization and actin cytoskeleton rearrangement, which collectively contribute to myogenic vasoconstriction (Fig. 2A) (Colinas et al., 2015).
Emphasis has also recently been placed on cell-to-cell junctions (Hill et al., 2009; Hill and Meininger, 2012; Schwartz, 2010). Cadherins, a family of Ca2+-dependent transmembrane proteins, involves cell-to-cell interactions which are involved in a number of biological processes including embryogenesis and tissue morphogenesis (George and Beeching, 2006; Jackson et al., 2010; Takeichi, 1991). It has been demonstrated that the intracellular domain of cadherins is coupled to catenin (a scaffolding protein) and the cadherin-catenin complex is provided for a nucleation site where actin cytoskeleton remodeling occurs (Aberle et al., 1996). N-cadherin has been shown to be the predominant cadherin expressed in rat resistance arterioles (Jackson et al., 2010; Jones et al., 2002). The question as to whether N-cadherin detects mechanical stresses on the vascular wall and initiates intracellular signaling for pressure-induced vasoconstriction has been approached in part using specific inhibitory antibodies or synthetic tripeptides (histidine-alanine-valine) for N-cadherin. Inhibition of N-cadherin markedly diminishes myogenic constriction, but not intracellular Ca2+ concentration, of rat cremaster arterioles (Jackson et al., 2010). The preceding investigations raise the possibility that N-cadherin may function as a part of the mechanosensory apparatus and be related to Ca2+ sensitization and/or cytoskeleton reorganization for the myogenic response. Indeed, it has recently been shown that adherens junctions formed by the novel mechanosensory N-cadherin in VSMC are involved in the myogenic response in superior cerebellar arteries (Sun et al., 2017). However, as precise downstream signaling pathways have not been fully investigated, mechanistic studies are required to further understand how N-cadherin contributes to pressure-induced vasoconstriction.
INVOLVEMENT OF MECHANOSENSITIVE GPCR IN MYOGENIC CONSTRICTION
The generally accepted concept of ligand-dependent activation of angiotensin II type 1 receptor (AT1R) has recently shifted to a new paradigm for mechanoactivation of the AT1R (i.e., ligand-independent activation) (Zou et al., 2004). There is mounting evidence showing that a number of GPCR possess inherent mechanosensitivity, thereby conceivably serving as primary biological mechanosensors that recognize membrane stretch or deformation of arteriolar myocytes caused by increased intraluminal pressure changes (Mederos y Schnitzler et al., 2008; Mederos y Schnitzler et al., 2011; Schleifenbaum et al., 2014; Storch et al., 2012; Storch et al., 2015; Yasuda et al., 2008). Supporting ligand-independent activation of GPCR, it was first demonstrated that mechanical activation of the AT1R leads to pressure overload-induced cardiac hypertrophy in the absence of angiotensin II production (Zou et al., 2004). This innovative study stimulated considerable interest in mechanisms by which the AT1R acts as a mechanosensor for initiating events associated with the myogenic response (e.g., sensation of membrane stretch or change of wall tension) and if mechanoactivation of the AT1R elicits downstream signaling pathways for pressure-induced vasoconstriction. In regard to this, it has been shown that angiotensin II-independent activation of the AT1R induced by hypotonic buffer-mediated cell swelling or positive pipette pressure increases cation influx through transient receptor potential canonical 6 channels (Mederos y Schnitzler et al., 2008). Transient receptor potential canonical 6 channels have been previously suggested to be involved in mechanosensation but are not themselves directly activated by membrane stretch (Mederos y Schnitzler et al., 2008). However, AT1R and transient receptor potential canonical 6-channel coexpression in human embryonic kidney cells results in increased cation current after application of mechanical stress (Mederos y Schnitzler et al., 2008). Thus, it is suggested that the AT1R is a key molecular mechanism for conversion of a mechanical stimulus into a biological signal.
Since the AT1R was identified to modulate the myogenic response, increasing emphasis has been placed on mechanistic studies relating to how mechanoactivation of the AT1R leads to myogenic constriction. It has been shown that osmotically induced membrane stretch of arteriolar myocytes reduces XE991-sensitive voltage-gated Kv channel currents, but the inhibited currents are not found in the absence of the AT1aR (Schleifenbaum et al., 2014). The AT1aR mechanoactivation-mediated suppression of K+ efflux via Kv channel may be coupled to membrane depolarization and myogenic constriction (Schleifenbaum et al., 2014). Surprisingly, in this study, interplay between the AT1R and the transient receptor potential canonical 6 channels was not observed, which is inconsistent with previous studies that suggested an important role for transient receptor potential canonical 6 channels in pressure-induced vasoconstriction (Mederos y Schnitzler et al., 2008; Voets and Nilius, 2009; Welsh et al., 2002). Subsequently, in arterioles of skeletal muscle that exhibits a relatively high level of myogenic tone under resting conditions, mechanoactivation of the AT1R in response to increased intraluminal pressure was also demonstrated to induce pressure-induced vasoconstriction (Hong et al., 2016). These studies further showed that the ligand-independent activation of the AT1R activated phospholipase C-mediated release of diacylglycerol with subsequent protein kinase C downstream signaling and actin cytoskeleton remodeling and in turn myogenic vasoconstriction (Fig. 2B). Moreover, another mechanism underlying AT1R-mediated myogenic constriction has been proposed in cerebral arteries (Gonzales et al., 2014). The mechanically activated AT1R is coupled to activation of Src tyrosine kinase that is necessary for phospholipase C gamma1-dependent generation of inositol trisphosphate (IP3) and diacylglycerol. Subsequently Ca2+ release from the sarcoplasmic reticulum (by IP3-mediated activation of IP3R and calcium-induced calcium release) evokes activation of transient receptor potential melastatin 4 channels, cation entry, membrane depolarization, and myogenic constriction (Fig. 2C) (Gonzales et al., 2014). Earley and colleagues then subsequently identified that the predominant AT1R interacts with transient receptor potential melastatin 4 channels for myogenic responsiveness in posterior cerebral arteries (Pires et al., 2017). However, it was reported in penetrating parenchymal cerebral arterioles that the expression level of AT1aR and AT1bR isotype is indistinguishable and AT1R contributes to initiating pressure-induced vasoconstriction, but not maintaining the myogenic reactivity, in the cerebral microvasculature (Yamasaki et al., 2020).
It is well reported that GPCR superfamily is diversely expressed in arteriolar myocytes. Thus, it is likely that myogenic vasoconstriction may occur following the activation of other putative mechanosensitive GPCRs. Indeed, a variety of receptors of endothelin, muscarinic, vasopressin, histamine, bradykinin, parathyroid hormone, dopamine, and purinergic P2Y4/P2Y6 have been appreciated as potential mechanoreceptors (Abdul-Majeed and Nauli, 2001; Brayden et al., 2013; Li et al., 2014; Mederos y Schnitzler et al., 2008; Storch et al., 2012; Zhang et al., 2009). Among them, purinergic receptors have received considerable attention. Myogenic constriction of pial arteries in the cerebral circulation is sensitive to treatment with candesartan (Brayden et al., 2013), indicating that the AT1R plays an important role in myogenic constriction of the cerebral resistance arteries. However, a similar contribution of the AT1R to myogenic response has not been identified in parenchymal cerebral arterioles, whereas purinergic P2Y4 or P2Y6 receptors appear mechanosensitive and contribute to myogenic constriction of the intracerebral vessels (i.e., parenchymal arterioles) in a manner of involving transient receptor potential melastatin 4-channel activation-dependent membrane depolarization (Brayden et al., 2013; Li et al., 2014). This suggests that pressure-induced vasoconstriction may be differentially initiated by disparate mechanosensitive GPCR in diverse vascular beds and/or species.
A novel GPCR mechanosensor, cysteinyl leukotriene 1 receptor, has been identified to participate in myogenic responsiveness (Storch et al., 2015). Activation of cysteinyl leukotriene 1 receptor has been traditionally reported to induce constriction of bronchial smooth muscle in asthma (Lynch et al., 1999; Singh et al., 2010). However, it has been found that pharmacological inhibition of cysteinyl leukotriene 1 receptor with either pranlukast or montelukast is surprisingly linked to reduced pressure-induced vasoconstriction of fourth-order mice mesenteric arterioles (Storch et al., 2015). Suppression of endogenous ligand leukotriene production using zileuton (a 5-lipoxygenase inhibitor) or a neutralizing leukotriene antibody does not influence on myogenic constriction, supporting that cysteinyl leukotriene 1 receptor is not activated by their ligand and may also play a crucial role in sensation of mechanical stresses in resistance arterioles.
One criterion for mechanoreceptors is that mechanical stimuli (i.e., tension, stretch, intraluminal pressure) are able to directly induce conformational changes of target mechanosensitive GPCR. Using a substituted cysteine accessibility mapping approach, a mechanically induced conformational change in the AT1R has been suggested. Importantly, this change in conformation is distinct from the ligand-dependent receptor conformation (Yasuda et al., 2008). Specifically these studies showed, direct stretch (20%) of cultured cells on extensible silicone dishes results in the seventh transmembrane protein of the AT1R rotating anticlockwise and dislocating towards the agonist-binding pocket of the receptor (Yasuda et al., 2008), whereas agonist-dependent activation of the AT1R correlates with the dislocation and rotation of the third transmembrane segment of the protein (Farrens et al., 1996; Karnik et al., 2003). Although there has not been direct evidence for the distinct conformational change of mechanically activated AT1R (e.g., crystallography studies), bioluminescence or fluorescence resonance energy transfer studies support that the AT1R favors a unique configuration (Mederos y Schnitzler et al., 2008; Rakesh et al., 2010). Therefore, the AT1R and other GPCR appear to have more than one active receptor conformation as shown in dynamic states of ion channels (Storch et al., 2012) and the distinct conformational changes in response ligand-mediated or mechanical stress-induced activation of the AT1R and other GPCR conceivably may confer the receptors to cause unique intracellular signaling and physiological outcomes.
An additional question relating to pressure/tension-induced activation of GPCRs is how does this mode of activation impact on their ability to signal in response to their classical ligand (i.e., in the case of the AT1R, angiotensin II). Does mechanoactivation affect the affinity of the receptor for its agonist or does the mechanical stimulus act as a ‘biased ligand’ (Wang et al., 2018) activating specific elements of the signaling pathways outlined in Fig. 1? These considerations are critical for understanding the interactions that occur between local and neuroendocrine mechanisms of blood flow control.
CONCLUSIONS
Small resistance arteries play a vital role in regulating blood flow locally in response to moment-to-moment changes in intravascular pressure. Physiologically, this intrinsic autoregulatory capacity of small arteries is required, in part, to match metabolic and hemodynamic demands in tissues and organs. Such responses are particularly well developed in metabolically active tissues including brain, heart, and skeletal muscle. Meanwhile, impaired autoregulation of small arteries may contribute to diverse pathologies, including vascular rupture, capillary damage, edema, vasospasms, ischemic stroke, or hypertension. Impaired local control of blood flow may also contribute to cardiovascular dysfunction in disorders such as diabetes. Thus, investigations into how biological mechanosensors detect the circumferential forces applied on the vascular wall and how this information is exactly transduced into signaling pathways for myogenic reactivity are of potential translational relevance in regard to developing strategies for the prevention and treatment of cardiovascular diseases that result from impaired pressure-induced autoregulation. It is increasingly realized that several cellular components on arteriolar smooth muscle cells (e.g., integrins, GPCR) act as mechanotransducers and thus potentially contribute to myogenic responsiveness. For a more complete understanding of the arteriolar myogenic response, further investigations are required to integrate these observations with other components of the overall myogenic signaling mechanism including extracellular matrix proteins, ion channels, Ca2+ handling, and contractile proteins.
ACKNOWLEDGMENTS
This work was supported by the Ministry of Education of the Republic of Korea and the National Research Foundation of Korea (NRF-2017S1A5B8067020).
Notes
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.