Aging, exercise, and extracellular matrix in the heart
Article information
Abstract
Aging is characterized by a progressive impairment of (a) cardiac structure including fibrosis and cardiomyocyte density, and (b) cardiac function including stroke volume, ejection fraction, and cardiac output. The cardiac remodeling involves loss of cardiac myocytes, reactive hypertrophy of the remaining cells, and increased extracellular matrix (ECM) and fibrosis in the aging heart, especially left ventricles. Fibrosis (i.e., accumulation of collagen) with aging is very critical in impairing cardiac function associated with increased myocardial stiffness. The balance of ECM remodeling via ECM synthesis and degradation is essential for normal cardiac structure and function. Thus an understanding of upstream ECM regulatory factors such as matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), and myofibroblasts is necessary for gaining new insights into managing cardiac remodeling and dysfunction with aging. In contrast, exercise training effectively improves cardiac function in both young and older individuals. Exercise training also improves maximal cardiovascular function by increasing stroke volume and cardiac output. However, limited data indicate that exercise training might attenuate collagen content and remodeling in the aging heart. We recently found that 12 weeks of exercise training protected against geometric changes of collagen ECM in the aging heart and ameliorated age-associated dysregulation of ECM in the heart, as indicated by up-regulation of active MMPs as well as down-regulation of TIMPs and TGF-β. This review will provide a summary and discussion of aging and exercise effects on fibrosis and upstream regulators of ECM in the heart.
INTRODUCTION
Myocardial tissue is composed of cardiac myocytes, nonmyocytes (e.g., fibroblasts, endothelial cells, vascular smooth muscle cells, etc), and extracellular matrix (ECM) proteins (Baudino et al., 2006; Bowers and Baudino, 2012; Camelliti et al., 2005; Curtis and Russell, 2011; Souders et al., 2009). Myocardial ECM is essential for proper cardiac structural integrity and pump function (Curtis and Russell, 2011). The ECM a) provides a scaffold for myocytes, fibroblasts, and endothelial cells, and b) transmits mechanical forces and signals to myocardial fibers (Baudino et al., 2006). The ECM also provides mechanical stability, physical strength, stiffness, ductility, and energy absorption to tissues. The ECM is essential for efficient cardiac function via myocyte alignment, regulating blood flow during contraction, and compliance. Moreover, the ECM is an important mediator of growth-related factor and in modulating the cardiac phenotype during development and hypertrophy. Therefore, the disruption of ECM homeostasis is a key factor for the progression of cardiac dysfunction (Baudino et al., 2006).
Myocardial ECM is composed of collagens (e.g., fibril-forming collagens and non-fibril forming collagens), glycoproteins (e.g., fibronectins, elastin, laminins, etc), proteoglycans, extracellular proteases, and ECM receptors (Corda et al., 2000; Goldsmith and Borg, 2002). ECM in the heart is linked to cellular cytoskeleton by transmembrane molecules, mainly integrins, which provides a physical connection between cytoskeleton and ECM proteins (Corda et al., 2000; Sarasa-Renedo and Chiquet, 2005). The interactions among ECM, cytoskeleton, and cell through integrins might be very important during cardiac remodeling (Goldsmith and Borg, 2002; Jane-Lise et al., 2000; Rosso et al., 2004). Although glycoproteins and proteoglycans are essential in proper cardiac geometry and various functions of the ECM, the most abundant structural components of the ECM are collagens (Bowers and Baudino, 2012), which are produced primarily by fibroblasts either on the membrane-bound ribosomes of the rough endoplasmic reticulum (ER) or placed within the ECM, respectively (Kjaer, 2004). The ability to synthesize the ECM components depends on cell types in the heart. For example, fibroblasts and smooth muscle cells synthesize collagen types I and III and fibronectin, whereas cardiac myocytes and endothelial cells produce collagen type IV (Corda et al., 2000). In addition, laminin is produced by cardiac myoctyes, smooth muscle cells, and endothelial cells (Corda et al., 2000). Alterations in the profile of ECM proteins can play a profound influence on the form and function of heart.
The aging heart is characterized by decreased myocyte number, increased myocyte size, and increased extracellular matrix compared with younger heart (Kwak et al., 2006). Cell death by apoptosis or necrosis is very critical determinant of ECM remodeling because it induces a loss of contractile tissue, reactive compensatory hypertrophy of remaining cardiomyocytes, and accumulation of collagen (i.e., fibrosis) and other ECM proteins (Jugdutt, 2003). These phenotypic changes of the myocardium with aging occur in the mainly left ventricle. For example, apoptosis, programmed cell death, is localized into the left ventricle, suggesting that it is initiated by mechanical factors (Kajstura et al., 1996). Overall, myocardial remodeling is determined by the consequence of changes in cardiac myocytes and disruption of ECM homeostasis. The ECM remodeling caused by aging results in myocardial remodeling, contributing to rearrangement of normally existing structures (Swynghedauw, 1999). The ECM remodeling also occurs in dilated cardiomyopathy (Pauschinger et al., 2002) and myocardial infarction (Lindsey et al., 2003). The ECM is a fibrillar network that embeds cardiomyocytes and the whole cardiac structure. The ECM remodeling is a critical part of mortality in the elderly. Furthermore, aging seriously affects the myocardial structure and function, as the fundamental biological process of aging is associated with an increased arterial hypertension, atherosclerosis, and decreased physical activity.
Fibrosis is a complicated tissue response that causes the excessive deposition of ECM, especially collagens (Krieg and LeRoy, 1998; Souders et al., 2009). Fibrosis, one of the major biological determinants of cardiac remodeling, is an increased collagen content and concentration, resulting in increased myocardial stiffness and cardiac dysfunction. Fibrosis is multifactorial, and it is resulted from aging, myocardial ischemia, inflammatory processes, hormones, vasoactive peptides, or diabetes (Swynghedauw, 1999). There are converging reports suggesting that myocardial fibrosis occurs in senescent hearts both in rats and humans (Kwak et al., 2011). Furthermore, there is emerging evidence that aging is associated with increased cardiac fibroblasts (Camelliti et al., 2005; Krenning et al., 2010). The ECM remodeling with aging including modifications of ECM protein synthesis and degradation would suggest that the aging heart might be unable to adapt to an increased load well (Burgess et al., 2001; Debessa et al., 2001; Mendes et al., 2012; Nguyen et al., 2001).
COLLAGENS IN THE HEART
Collagens are a regulated family of ECM proteins that provide structure and optimize function of the heart (Baudino et al., 2006; Souders et al., 2009). Presently, more than 20 collagen types have been identified in various vertebrate tissues. Collagen is the most abundant protein in ECM and forms the essential mechanical building blocks, providing tensile strength and resisting stretch (Jugdutt, 2003). The collagen is composed of three α-chains called triple helix or tropocollagen molecule. The common structure of collagens is repeating amino acid sequence (Gly-X-Y) that comprises the collagen chain. Most of collagens are present in the forms of polypeptide chains called collagen molecule or α-chain, consisting of glycine, proline, and hydroxyproline with hydroxylysine (Jugdutt, 2003). The network of collagens structurally exists at three levels named a) endomysium surrounding individual muscle fibers, b) perimysium surrounding groups of myocytes, and c) epimysium surrounding the entire muscle (DeSouza, 2002). Connective tissue consists mainly of collagen, and to a much lesser extent, fibronectin, laminin, and elastic fibers (Carvalho Filho et al., 1996).
The collagens can be divided into two major classes, the fibrillar-forming and non-fibrillar-forming collagens (Jugdutt, 2003). Among collagens, five of collagens (I, II, III, V, and XI) form fibrils (Jugdutt, 2003). The fibril-forming collagens provide the structural framework of tissues. In particular, collagen types I and III in myocardial collagens are predominantly interstitial collagens in the heart that surround cardiac myocytes and the coronary microcirculation, providing structural integrity for the cardiomyocytes (Goldsmith and Borg, 2002; Jugdutt, 2003; Kassiri and Khokha, 2005). Type I collagen type makes up approximately 85% and type III collagen 11% of total collagen in the heart (DeSouza, 2002; Jugdutt, 2003). Although collagen types I and III coexist in the ECM, especially both in the perimysium and endomysium (DeSouza, 2002), there are some differences due to the composition of the α-chains that comprise the collagen triple-helix. Collagen type I is a hybrid, consisting of two identical α1 (I) chains and one α2 (I) chain that form the superhelix (Debessa et al., 2001). Collagen type I is thick, yellow or red, strong fibers and is thought to play an essential role in providing structural stability to tissues, whereas collagen type III contains three identical α1 (III) chains (Debessa et al., 2001). Collagen type III is thin, greenish fibers, and fine reticular network in most soft connective tissue unlike the larger fibers that are derived from collagen type I molecules (Debessa et al., 2001). Collagen type I provides high tensile strength and stiffness to tissues, whereas collagen type III provides high compliance to tissues (Jugdutt, 2003). So, the ratio of collagen type III to I has been implicated in functional properties of the heart, with a higher ratio of collagen type III to I indicating more compliant tissue and a lower ratio of collagen type III to I indicating a stiffer, less compliant tissue (Jugdutt, 2003).
REGULATION OF CARDIAC COLLAGEN ECM
Collagen ECM plays an important role in cardiovascular function, and remodeling in the ECM contributes to myocardial dys-function (Goldsmith and Borg, 2002; Porter and Turner, 2009). Myocardial failure and remodeling are usually characterized by collagen accumulation, collagen fibril disruption, myocyte loss via apoptosis or necrosis, and impaired rearrangement of structure (Swynghedauw, 1999). In particular, accumulation of collagen ECM with aging in the heart could create a mechanical environment and stress distribution that contributes diminished systolic performance, decreased compliance, and diastolic dysfunction (DeSouza, 2002). Therefore, the balance of ECM remodeling via collagen ECM synthesis and degradation is essential for normal cardiac structure and function (Jugdutt, 2003; Souders et al., 2009). Collagen ECM remodeling is modulated by regulatory proteins, hormonal factors, cytokines, and growth factors (Baudino et al., 2006; Camelliti et al., 2005). Thus an understanding of upstream ECM regulatory factors such as MMPs, TIMPs, TNF-α, TGF-β, and myofibroblasts provides therapeutic strategies to protect against cardiac remodeling and dysfunction with aging (Fig. 1).
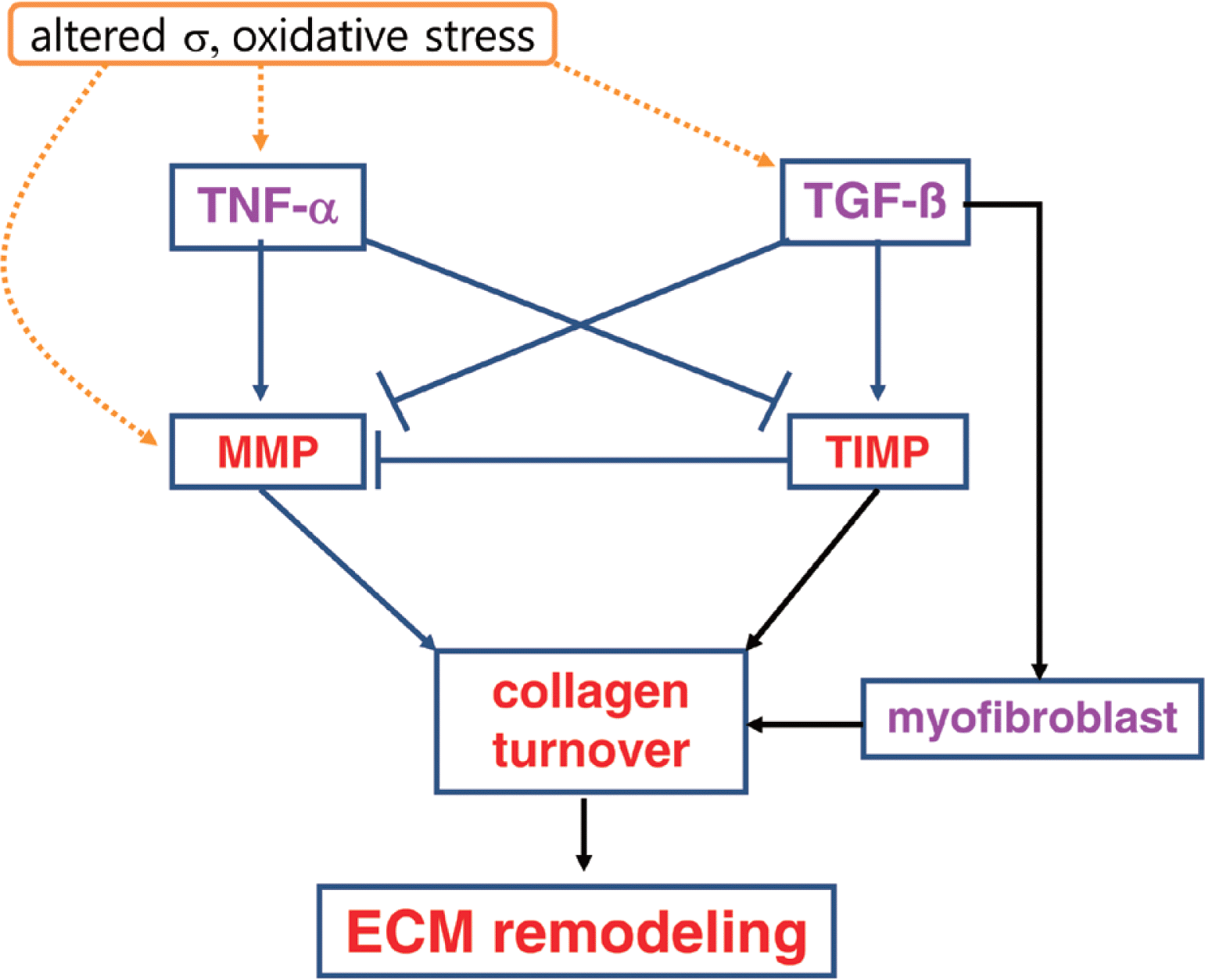
Collagen ECM turnover signaling in the heart. Altered mechanical stress (σ) and oxidative stress may stimulate TNF-α, TGF-β, and MMP. TNF-α may stimulate MMP and inhibit TIMP. However, TGF-β may inhibit MMP and stimulate TIMP and myofibroblast. Finally, MMP degrades collagens, but TIMP and myofibroblast inhibit collagen degradation and promote collagen synthesis, which determine collagen ECM remodeling.
Cardiac MMPs and TIMPs
ECM depends on a balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), which determines cardiac remodeling (Ahmed et al., 2006; Jugdutt, 2003; Benjamin and Khalil, 2012). MMPs are an endogenous family of enzymes that degrade ECM proteins, which are responsible for ECM remodeling in a number of physiological and pathological process (Ahmed et al., 2006; Benjamin and Khalil, 2012; Tsuruda et al., 2004). To date, the MMP family consists of more than 20 unique proteins in vertebrates. Most of MMPs are inactive enzymes that are activated in ECM. It has been shown that MMPs highly related with myocardial remodeling are collagenases (e.g., MMP-1 and MMP-13), gelatinases (e.g., MMP-2 and MMP-9), stromelysin (e.g., MMP-3), and the membrane-type MMP (e.g., MMP-14) (Kassiri and Khokha, 2005). These kinds of MMPs degrade predominantly collagen types I and III in the ECM of the heart (Schupp et al., 2006).
MMPs are Ca2+-and Zn2+-dependent proteases that are usually synthesized as an inactive form or pro-MMP, which is activated by the cleavage of an amino-terminal propeptide domain either by autoproteolysis, another MMP, or serine protease (Jugdutt, 2003). For example, MMP-14 activates MMP-2, which requires TIMP-2 binding to its active place (Lafleur et al., 2003). MMPs in the heart are expressed primarily by fibroblasts (Chapman et al., 2003) and cardiomyocytes (Coker et al., 1999). Most pro-MMPs are stored extracellular bound to different ECM components. Upon stimulation, activated MMPs degrade the ECM proteins including collagens, fibronectin, laminin, gelatin, and proteoglycan. Therefore, MMPs are significant regulators of ECM turnover in the heart, thus contributing to physiological function as well as pathology. In contrast, activity of MMPs is in part regulated by endogenous inhibitors (Benjamin and Khalil, 2012; Jugdutt, 2003). TIMPs are specific MMP inhibitors in the ECM (Lovelock et al., 2005). The role of TIMPs is to prevent excessive ECM degradation by MMPs. There are 4 TIMPs identified in vertebrates, TIMP-1, -2, -3, and -4, acting as the natural inhibitors of active forms of all MMPs through binding to MMPs in a 1:1 ratio (Cleutjens and Creemers E, 2002). Among them, TIMP-1, -2, and -4 are soluble forms, whereas TIMP-3 binds to the ECM via heparan sulfate proteoglycans within the ECM (Yu et al., 2000). The balance between MMPs and TIMPs plays a critical role in the process of cardiac ECM remodeling which contributes to cardiac function. Based on previous findings, it appears that cardiac ECM remodeling is generally associated with enhanced MMP and reduced TIMP activities (Jugdutt, 2003). However, there are differences in the studies. For example, the levels of TIMP-1 were either repressed (Tyagi et al., 1996) or increased (Thomas et al., 1998) in dilated cardiomyopathy patients.
It has been shown that inhibition of MMP activity is beneficial during cardiac remodeling and wall stress following injury due to myocardial infarction (Benjamin and Khalil, 2012; Peterson, 2006). For example, Rohde et al. (1999) indicated that a broad range MMP inhibitor attenuated left ventricular dilatation 4 days after infarction in a mouse myocardial infarction. In addition, inhibition of MMP-9 activity attenuated left ventricular enlargement and collagen content after myocardial infarction (Ducharme et al., 2000). Therefore, MMP inhibition might be a new therapeutic treatment to control cardiac dysfunction and failure. A few publications indicated that MMP levels increased, and TIMP levels decreased in the rat heart with advancing age. For example, Lindsey et al. (2005) found that the levels of MMP-3, MMP-8, MMP-9, MMP-12, and MMP-14 increased, and the levels of TIMP-3 and TIMP-4 decreased in the insoluble fraction of old mice, compared with young adult mice, suggesting that aging is associated with increased ECM degradative capacity. However, much different findings were reported by Robert et al. (1997). Their results indicated a 40–45% decrease in both MMP-2 and pro-MMP-1 activity and mRNA in 24-month-old rat heart, suggesting that the reduction of ECM degradation pathway by MMP allows accumulation of collagen and promotion of age-associated fibrosis. Thus, the current literature is unclear about MMP or TIMP expression with aging in the heart.
Cardiac TNF-α
Upstream regulators of MMPs and TIMPs include inflammatory cytokines in the heart (Siwik and Colucci, 2004; Tsuruda et al., 2004). It seems likely that the cytokines may lead to an imbalance in myocardial MMP/TIMP ratio resulting in altered myocardial ECM architecture and development of left ventricle remodeling and dysfunction (Jugdutt, 2003; Siwik and Colucci, 2004). Among cytokines, tumor necrosis factor-α (TNF-α), a pro-inflammatory cytokine, can increase the matrix collagen degradation by upregulating MMP activity and downregulating TIMPs (Jugdutt, 2003; Murray et al., 2010; Siwik and Colucci, 2004). TNF-α has a variety of different biological capacities in response to one or more different forms of environmental stress in heart failure, including LV dysfunction, cardiomyopathy, LV remodeling, abnormalities of mitochondrial energetics, increased production of reactive oxygen, and cardiac myocyte apoptosis (Mann, 2002). In particular, LV remodeling by TNF-α is involved in alterations in the biology of the cardiac myocyte, progressive myocyte loss, and alterations in ECM including synthesis and degradation of collagen matrix (Mann, 2002; Murray et al., 2010).
Significantly increased levels of TNF-α have been demonstrated in patients with dilated or ischemic cardiomyopathy (Oral et al., 1999) and in animal models of myocardial infarction (Irwin et al., 1999). Furthermore, Li et al. (2000) suggested that cardiac over-expression of TNF-α in transgenic mice caused increases in MMP-2 and MMP-9 activity as well as marked diastolic dysfunction. In isolated cardiac fibroblasts, TNF-α decreases collagen synthesis, increases MMP expression, and decreases TIMP expression (Siwik et al., 2000). In contrast, Sivasubramanian et al. (2001) reported that there were significant decreases in total MMP activity and elevated TIMP-1 levels in the cardiac overexpression of TNF-α in transgenic mice, suggesting a possible mechanism for the increase in myocardial fibrosis. Mann (2002) also showed that TNF-α promoted cardiac fibroblast proliferation and fibrosis. Although the mechanisms by which TNF-α affect MMP and TIMP may depend on in vitro and in vivo models, TNF-α may indeed induce an imbalance in MMP/TIMP ratio, remodeling and fibrosis in the heart.
Cardiac TGF-β
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that plays an important role in cell migration, proliferation, differentiation, apoptosis, and ECM protein production (Annes et al., 2003; Baudino et al., 2006; Hinck, 2012; Sales et al., 2006). TGF-β, an anti-inflammatory cytokine, is a potent stimulator of collagen synthesis (Siwik and Colucci, 2004). It consists of three isoforms, TGF-β1, TGF-β2, and TGF-β3 that are structurally and functionally closely related to one another (Annes et al., 2003). The TGF-β released from platelets and leukocytes stimulates the synthesis of ECM components including collagens, fibronectin, proteoglycans, and integrins in tissue repair after injury. It mediates collagen synthesis through increasing transcription and decreasing collagen degradation via reduced MMPs or enhanced TIMPs, thus favoring an accumulation of ECM and especially of collagen (Siwik and Colucci, 2004). For example, Seeland et al. (2002) suggested that the overexpression of TGF-β1 in transgenic mice resulted in increased protein expression of collagen types I and III, reduced interstitial collagenase protein activity and mRNA expression, and increased TIMP-1, -2, and -4 protein levels in the heart. Additionally, in cardiac fibroblasts, procollagen formation was stimulated by mechanical loading and TGF-β.
Acute exercise or mechanical loading may stimulate TGF-β synthesis in the heart (Calderone et al., 2001), smooth muscle (Gutierrez and Perr, 1999), skeletal muscle (Gavin and Wagner, 2001), and circulating blood (Heinemeier et al., 2003) as a physiological response. For example, Calderone et al. (2001) reported that TGF-β1 mRNA increased in the left ventricle of a voluntary exercise rat model of physiological cardiac hypertrophy. However, excessive and chronic expression of TGF-β is associated with many fibrotic diseases including cardiac fibrosis after infarction, lung fibrosis, and scarring (Annes et al., 2003). TGF-β may play a role in stimulating abnormal accumulation signaling of ECM proteins in the cardiovascular diseases. Rosenkranz et al. (2002) showed that TGF-β overexpression in the transgenic mice heart resulted in cardiac hypertrophy and fibrosis. Similarly, Brooks and Conrad (2000) found that TGF-β1 deficient old mice heart exhibited a decrease in myocardial fibrosis and reduced myocardial stiffness, indicating the role of TGF-β to contribute to ECM component synthesis in the heart.
Cardiac myofibroblasts
Myofibroblast is a differentiated cell type from fibroblast characterized by increased ECM protein synthesis called fibrosis formation, providing an essential role for ECM remodeling during normal and pathological wound healing (Cleutjens and Creemers, 2002; Powell et al., 1999; Tomasek et al., 2002). Myofibroblasts as a smooth-muscle like fibroblasts might be produced from progenitor stem cells in the heart or from the circulation, and secret cytokines (e.g., TNF-α), growth factors (e.g., TGF-β), chemokines, and inflammatory mediators (Porter and Turner, 2009; Powell et al., 1999). In addition, differentiation to the myofibroblast may be induced by transforming growth factor-β1 (TGF-β1) (Tomasek et al., 2002). Myofibroblast expression may be not detectable in the normal healthy adult hearts, while myofibroblasts are often associated with injured heart such as myocardial infarction for wound healing (Baudino et al., 2006; Poobalarahi et al., 2006). In particular, Poobalarahi et al. (2006) reported that increased type I collagen synthesis by myofibroblasts was accompanied by a significant increase in collagen deposition into insoluble ECM in the heart. Accordingly, myofibroblasts appear to play a critical role in production of cardiac ECM in response to injury (Porter and Turner, 2009). In addition, differentiated myofibroblasts are unique in that they express α-smooth muscle actin (α-SMA) unlike adult fibroblasts (Chaponnier and Gabbiani, 2004; Powell et al., 1999).
Expression of α-SMA positive myofibroblasts appears to be regulated by TGF-β1 (Chaponnier and Gabbiani, 2004). A similar finding was also reported that TGF-β1 promoted the conversion of myofibroblasts in vitro (Gabbiani, 2003). Additionally, Kuwahara et al. (2002) found that TGF-β1 function-blocking antibodies administered to pressure-overload rats prevented the myofibroblasts conversion in cardiac interstitium and subsequent increases in mRNA of type I collagen as well as diastolic heart failure. Interestingly, Porter et al. (2004) showed that TNF-α via a TNF-R1 receptor also increased myofibroblast proliferation in human heart.
AGING, EXERCISE, AND COLLAGEN ECM IN THE HEART
Aging and cardiac collagens
Myocardial remodeling during aging is related with changes in the amount and organization of ECM components (Swynghedauw, 1999; Kwak et al., 2011). In particular, myocardial collagens in ECM undergo remodeling with aging. A healthy arrangement of collagens provides a framework for myocyte sheath sliding, transmittance of force from myocyte to the ventricular chamber, prevents excessive stretch and damage, and preserves heart function (DeSouza, 2002). However, excessive accumulation of collagen matrix is up-regulated in a number of cardiovascular diseases. Moreover, aging also increases the rate of ventricular collagen turnover and deposition by fibroblasts called fibrosis (Baudino et al., 2006; Mendes et al., 2012; Thomas et al., 2000, 2001). Fibrosis with aging is characterized by increased collagen content (Hwang et al., 2007), decreased collagen solubility, and increased collagen cross-linking (Thomas et al., 2000, 2001). This increase in collagen deposition during aging may be thought to result from a combination of cellular events including increased collagen synthesis and decreased degradation (Kwak et al., 2011). The collagen might become more resistant to collagenase degradation with aging (Jugdutt, 2003). Excessive accumulation of collagen in the heart could lead to tissue stiffness, increase the incidence of arrhythmias, disrupt electronic communication between myocytes, and result in diastolic and systolic dysfunction and heart failure (Baudino et al., 2006; Mendes et al., 2012).
Previous studies have demonstrated age-related changes in cardiac collagen concentration (Hwang et al., 2007; Lindsey et al., 2005; Mendes et al., 2012; Nguyen et al., 2001; Thomas et al., 2000, 2001). Debessa et al. (2001) indicated that the number and thickness of Type I collagen increased from adulthood to old age in human heart. Studies in animal hearts also provided consistent evidence of an increase in myocardial collagen concentration with aging (Nguyen et al., 2001; Thomas et al., 2000, 2001). These findings were confirmed by Nguyen et al. (2001), who examined the collagen concentration in the left ventricles of Fischer 344 rats at 6, 18, and 24 months of age. Their results revealed that the collagen concentration, as determined by hydroxyproline assay, progressively increased during aging with greatest increments from 6 to 18 months, then leveling off at 24 months. Similar findings were previously described by Mays et al. (1988), who found a gradual increase in collagen concentration, based on hydroxyproline levels between 2 weeks and 24 months of age. In addition, Lindsey et al. (2005) reported that total collagen levels increased with advancing age. Taken together, the increased collagen content/ concentration might be an integral part of ECM remodeling that takes place in the left ventricle consequent to the natural aging process leading to an increase in myocardial passive stiffness and impaired contractile function.
A few studies also have showed age-related increases in collagen cross-linking in cardiac muscle (Thomas et al., 2000, 2001). Increased collagen cross-linking could be implicated as a potential mechanism for an impaired extensibility and increased stiffness in aged heart. For example, Thomas et al. (2000, 2001) reported that there were significant overall age–related increases in collagen cross-linking in the both left ventricle and septum. In addition to the heart, skeletal muscle also showed the same phenomena that collagen content and collagen cross-linking significantly increased from young to senescence in skeletal muscle (Gosselin et al., 1998).
Exercise and cardiac collagens
Alterations in collagen profile have been shown to occur following exercise training in heart (Thomas et al., 2000, 2001) and skeletal muscle (Gosselin et al., 1998). For example, Thomas et al. (2001) observed that ten weeks of treadmill exercise training reduced age-induced up-regulation of collagen concentration (percent collagen) in the left ventricle septum of rats. The collagen cross-linking (HP) of left ventricle free wall was significantly lower in old trained rats, compared with their sedentary counterparts (Thomas et al., 2000, 2001). In addition, our research group recently demonstrated that 12 weeks of exercise training in rats significantly ameliorated age-associated increases in extramyocyte space and collagen-positive staining (Kwak et al., 2006, 2011). We also found that exercise training protected against age-related down-regulation of active MMPs (e.g., MMP-1, MMP-2, MMP-3, and MMP-14). Consistent with the inhibitory effects of TIMP-1 on MMP activation, aging dramatically increased TIMP-1 protein levels, whiles exercise training alleviated age-induced increase in TIMP-1 protein levels (Fig. 2). Exercise training also mitigated age-associated increase of TGF-β1 in the heart (Fig. 2). TGF-β1 is a potent stimulator of TIMP-1 and a potential contributor to fibrosis in the aging heart.
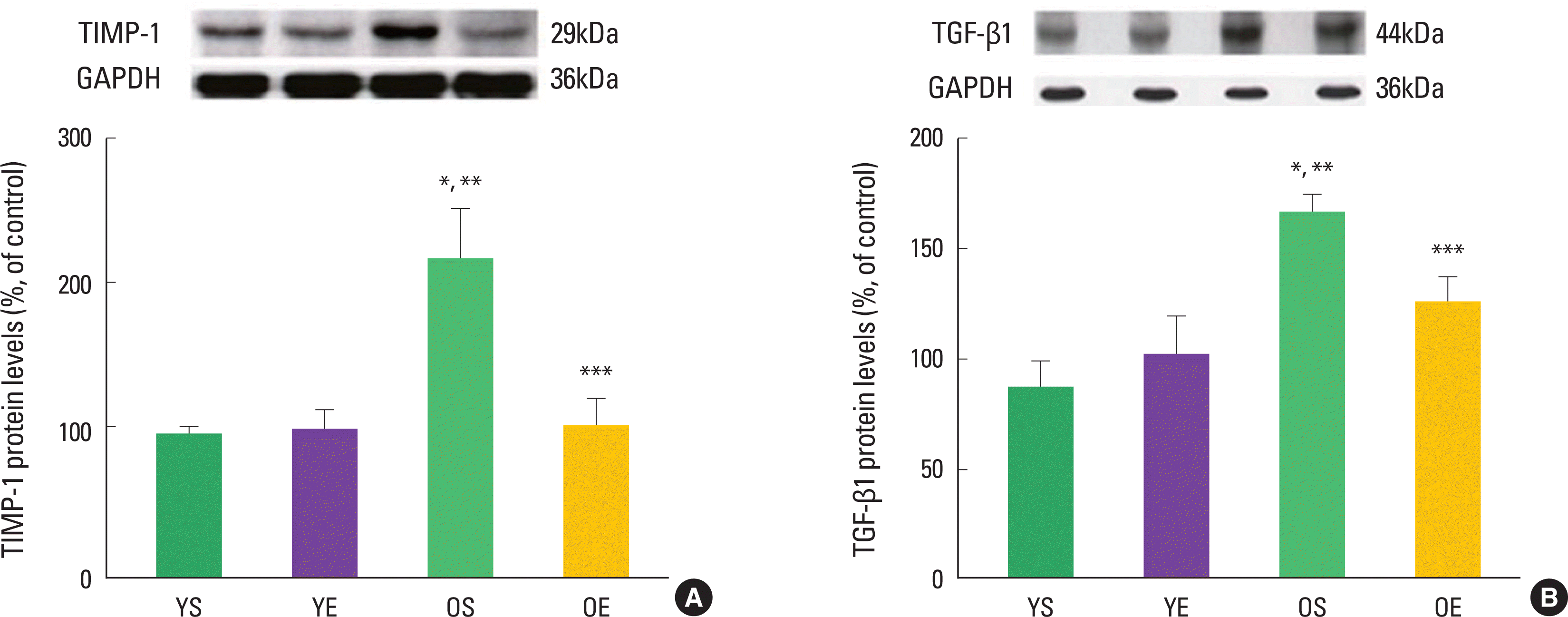
Effects of aging and exercise training on TIMP-1 and TGF-β1 in the heart. (A) TIMP-1 protein levels and (B) TGF-β1 protein levels of left ventricles in young sedentary (YS), young exercise (YE), old sedentary (OS), and old exercise (OE) groups (Kwak et al., 2011).
Conflicting results were also reported by others. Burgess et al. (1996) suggested that total collagen concentration (hydroxylproline) of rat left ventricle did not change by 10 weeks of treadmill exercise training. In addition, collagen type III -to-I ratio was not altered by exercise training in the rat heart (Burgess et al., 1996). In addition, Woodiwiss et al. (1998) found that 16 weeks of habitual voluntary wheel running had no effects on myocardial collagen concentration and cross-linking in the rat left ventricle, although cardiac stiffness was reduced. Similarly, Jin et al. (2000) showed that mRNA levels of collagen types I and III did not change with 13 week treadmill exercise training in the rat heart. So, based on previous findings, the role of exercise training on collagen concentration and cross-linking in the heart remains to be clarified.
CONCLUSIONS
In summary, aging resulted in increases in extramyocyte space and collagen contents associated with down-regulation of MMPs and up-regulation of TIMP-1 and TGF-β1 in the heart. However, exercise training ameliorated the age-related alterations in pathway signaling (e.g., MMPs-TIMP-1-TGF-β1), suggesting that exercise training protects against fibrosis and ECM remodeling in the aging heart. Further research is necessary to identify therapeutic targets to mitigate fibrosis, cardiovascular disease, and heart failure prevalent with advancing age.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1A1042383) and Inha University Research Grant (INHA-47286)
Notes
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.