INTRODUCTION
The term of “psychoneuroimmunology” was introduced in 1960s in order to define the bidirectional relationship between brain and immune system (Straub and Cutolo, 2018). In this respect, there have been increasing interests in the possible involvement of the immune system in psychiatric disorders. Moreover, changes in the immune system can lead to profound alterations in psychological state such as mood and behavior. The most possible link interacting behavior, nervous, endocrine and immune systems are established by the cytokines which act as signaling molecules of the immune system. Several studies indicated a close association between activation of pro-inflammatory cytokines including interleukin-1 (IL-1), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) and psychiatric symptoms (Anisman et al., 2005; Arisi, 2014; Konsman et al., 2002; Levine et al., 1999). During systemic infections, cancer or autoimmune diseases, pro-inflammatory cytokines can induce behavioral symptoms referred to as sickness behavior which is accompanied by a feeling of helplessness, depression, anxiety, excessive sleep, decreased appetite, and decreased concentration (Dantzer, 2001). These studies suggest that cytokines play an important role in the mood disorder and behavioral dysfunction.
In the clinical study, the increase in plasma concentrations of the pro-inflammatory cytokines observed in patients suffering from depression is known to be closely correlated with the severity of this psychiatric disorder, the hyperactivity of the hypothalamic- pituitary-adrenal (HPA) axis and the deficiency in serotonergic (5-HT) neurotransmission (Kim et al., 2002; Maes et al., 1993; Maes et al., 1995; Maes et al., 1997; Maes et al., 1999; Nunes et al., 2002). Several medical conditions e.g., autoimmune disease such as rheumatoid arthritis, cancer, or hepatitis C therapies producing activation of the immune system are associated with psychological and neuroendocrine changes that resemble the characteristics of depression including depressed mood, appetite disturbance, sleep disturbance, psycho motor disturbance, fatigue, loss of energy, difficulty in thinking or concentrating, activation of the immune system, hyperactivity of HPA axis, and monoaminergic alterations (Bonaccorso et al., 2000; Rogler and Andus, 1998; Van Gool et al., 1999; Weinblatt et al., 1999). Similarly, systemic administration of pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α in animals induces increase HPA activity (Dantzer, 2001; Dorr, 1993; Yirmiya, 2000; Yirmiya et al., 2000). Recently, evidence supports a role of cytokines, especially IL-1, IL-6, TNF-α, and IFN-γ in the pathophysiology of depression and anxiety (Dorr, 1993; Guo et al., 2014). This could be due to the actions of pro-inflammatory cytokines as signaling molecules on neurotransmitter metabolism and neurocircuitry in the brain. This review focuses on the possible influences of neuroinflammation which may cause pathogenesis of affective disorders including depression and anxiety (Fig. 1).
CYTOKINES
Cytokines are signaling molecules released from the periphery immune cells (monocytes, macrophages, and lymphocytes). They act as intercellular messengers and are associated with activation of the immune system, cell differentiation, and cell death (Allan and Rothwell, 2003; Salim et al., 2012). Cytokines can be divided into pro-inflammatory cytokines such as IL-1β, IL-6, IFN-γ, and TNF-α which activate inflammation. In contrast, anti-inflammatory cytokines such as interleukin-4 (IL-4), interleukin-10 (IL-10), and interleukin-13 (IL-13) inhibit inflammation (Abbas et al., 1996). Cytokines interact with each other to keep the balance between pro-inflammatory and anti-inflammatory cytokines. Systemic injection of pro-inflammatory cytokines induces depression- like behaviors (Dantzer et al., 2001), whereas treatment of antidepressants in depression attenuates cytokine production and their action (Barden, 1999; Castanon et al., 2002; Schiepers et al., 2005). Pro-inflammatory cytokines produced from the peripheral immune system are delivered directly to the central nervous system or indirectly affect the brain by increasing nitric oxide (NO), prostaglandins (PGE2) (Capuron and Miller, 2011). Cytokines are also produced by neurons and glial cells (microglia, astrocytes, and oligodendrocytes) of the brain and can cause or reflect abnormalities in the central nervous system (Becher et al., 2017).
NEUROINFLAMMATION
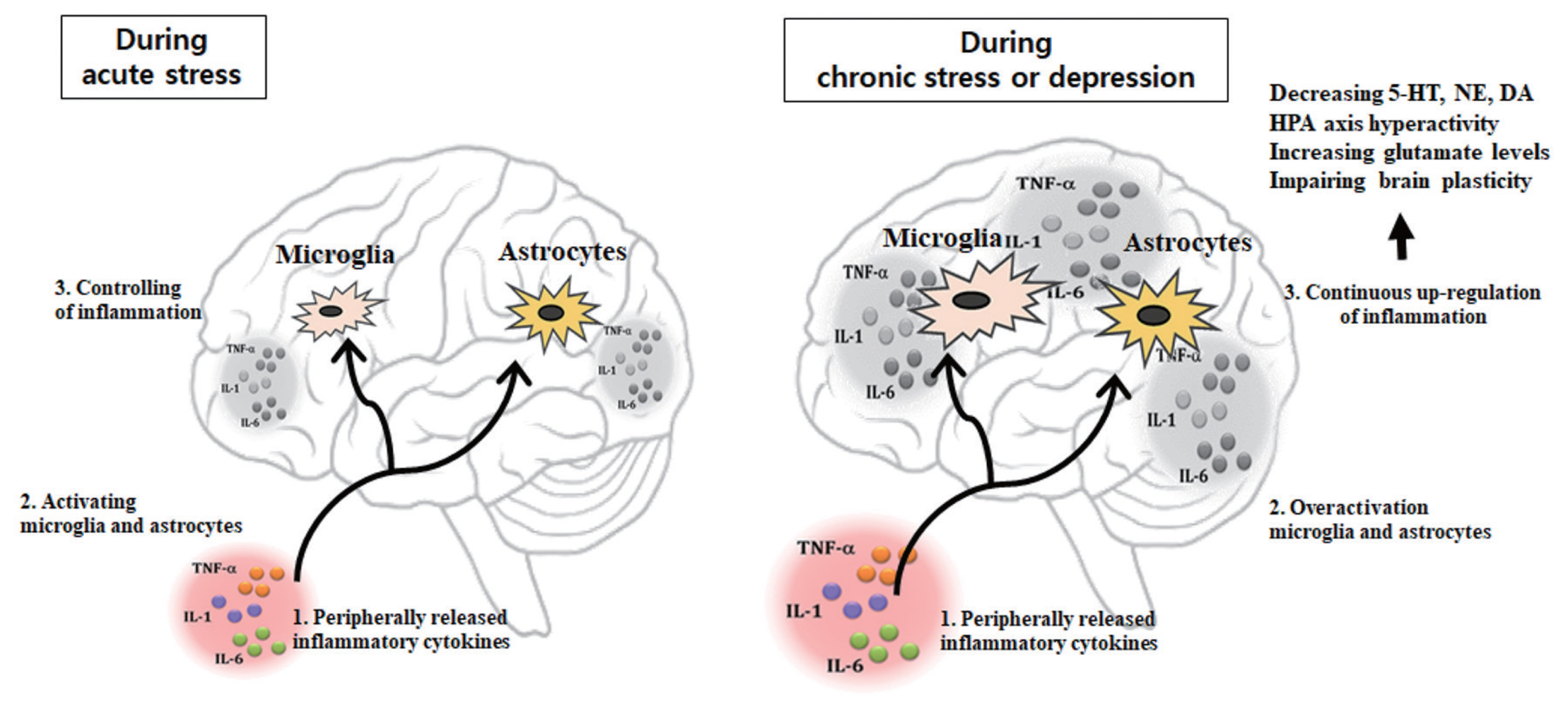
Peripheral cytokines are hydrophilic and because of their relatively high weight they cannot pass through blood-brain barrier (BBB) under normal conditions. However, in pathological conditions with increased BBB permeability, peripheral cytokines can pass through the BBB (Lacroix and Rivest, 1998; Wong et al., 1996). Also, peripherally produced cytokines can cross the circumventricular organs and the choroid plexus where relatively leaky regions of the BBB (Quan and Banks, 2007). Another mechanism is the binding of cytokines to peripheral afferent nerve fibers in the vagus nerve then relay cytokine signals to relevant brain regions through activation of the nucleus of the tractus solitarius and area postrema (Capuron and Miller, 2011; Miller et al., 2009). In then, transmission of these cytokine signals occurs through secondary mediators, PGE2, that elicit physiological and behavioral responses. Peripherally activated cytokines are actively transported into the brain-stimulating microglial and astrocytes cells, which in turn produce cytokines (Müller and Ackenheil, 1998). Microglia which is the principal cellular mediators of inflammatory processes in the brain (Graeber, 2010; Wake et al., 2009) can produce pro-inflammatory cytokines, NO, and reactive oxygen species (Harry and Kraft, 2008).
Furthermore, increased microglia by peripheral inflammation recruits monocytes into the brain and the cerebral infiltrated monocytes can further produce pro-inflammatory cytokines (D’Mello et al., 2009; Peraçoli et al., 2003). Postmortem and positron emission tomography (PET) imaging studies identified microglia activation in individuals who committed suicide (Schnieder et al., 2014; Steiner et al., 2008; Torres-Platas et al., 2014). Such increases in pro-inflammatory cytokines may influence the brain by increasing neurotoxic metabolites, or directly exerting neurotoxic effects on specific brain regions implicated in regulation of emotional response, including the amygdala, hippocampus, hypothalamus, and the cerebral cortex (Shim et al., 2019). Moreover, cytokine signaling in the brain is known to regulate important brain functions including neurotransmitter metabolism, neuroendocrine function, and mood-regulating neurocircuitry. The outcome of any type of dysregulation of the immune system in the brain might lead to occurrence of depression and anxiety (Salim et al., 2012). Inflammatory cytokines such as TNF-α and IL-1β may also activate microglia, reducing neuroplasticity and consequently depressing the function of neuronal circuits which is involved in mood regulation (Miller and Raison, 2016). Increased IL-1β activates astrocytes (Czéh et al., 2006) and some studies reveal an influence of different classes of antidepressant medications on astrocytes (Banasr et al., 2010; Rajkowska and Stockmeier, 2013; Wang et al., 2014). Astrocytes regulate synaptic and intracellular levels of neurotransmitters including glutamate and γ-aminobutyric acid (GABA) via transporters located on their processes (Lee et al., 2011; Sattler and Rothstein, 2006; Sofroniew and Vinters, 2010). In laboratory animal study, hypothalamic astrocytes activation following IL-1β infusion induced anxiety symptoms and hypothalamic astrocytes inhibition reduced anxiety behaviors which were relevant with astrocytic GABA release (Shim et al., 2019).
EFFECTS OF NEUROINFLAMMATION ON NEUROTRANSMITTERS
Pro-inflammatory cytokines including IL-1β, IFN, and TNF can reduce the bioavailability of neurotransmitters such as serotonin (5-HT), norepinephrine (NE), dopamine (DA) and excitatory amino acid glutamate (Miller and Timmie, 2009). Increased pro-inflammatory cytokines may reduce 5-HT availability in the synapse by activating of indolamine 2,3 dioxygenase (IDO), the enzyme to metabolize tryptophan which is the primary amino acid precursor of 5-HT, into kynurenine resulting in decreased 5-HT synthesis (Capuron et al., 2002; Capuron et al., 2003). In addition, activation of p38 mitogen-activated protein kinase can upregulate the expression and activity of the membrane transporter for 5-HT, NE, and DA (Miller and Timmie, 2009). Cytokine-induced activation of NO can lead to decreased tetrahydrobiopterin which serves as a coenzyme for tyrosine hydroxylase, the rate- limiting enzyme in DA synthesis in leading to decreased DA as well as NE availability. Several studies have documented that activation of IDO in the brain plays a critical role in the development of depressive-like behavior in rodents (Lestage et al., 2002; O’Connor et al., 2009). Moreover, increases in kynurenine and decreases in tryptophan levels have been associated with the depression and severity of depressive symptoms in humans (Capuron et al., 2003). Activated microglia can convert kynurenine into quinolinic acid (QUIN), which is a potent N-methyl-d-aspartate (NMDA) receptor agonist, a glutamate receptor, and together with cytokine-induced reduction in astrocytic glutamate reuptake and stimulation of astrocyte glutamate release, which in turn can lead to excessive glutamate. Excessive glutamate, especially when binding to extra synaptic NMDA receptor, can, in turn, lead to decreased brain-derived neurotrophic factor (BDNF) and excite toxicity leading to depression (Harry and Kraft, 2012; Hashimoto et al., 2013).
EFFECTS OF NEUROINFLAMMATION ON NEUROCIRCUITRY
To study microglial activation in depression in vivo, PET imaging studies using various PET ligands for the microglial marker translocator protein have been conducted. In human postmortem studies, Depression associated microglial activation is particularly observed in the prefrontal cortex, insula, and anterior cingulate cortex (ACC) have been correlated with the severity and duration of depression (Schnieder et al., 2014; Setiawan et al., 2018; Steiner et al., 2008). In addition, suicide has been related not only to microglial activation, but also to an enhanced perivascular macrophage around blood vessels (Torres-Platas et al., 2014). Inflammation can affect growth factors such as BDNF in the dentate gyrus of the hippocampus, and also affect fundamental aspects of neuronal integrity including neurogenesis, long-term potentiation, and dendritic sprouting, ultimately affecting learning and memory. Cytokine may influence neurotransmitter systems, especially DA, resulting in inhibiting several aspects of reward motivation and anhedonia in corticostriatal circuits involving the basal ganglia, ventromedial prefrontal cortex and subgenual and dorsal ACC (sgACC and dACC, respectively), while also activating circuits regulating anxiety, arousal, and fear including the amygdala, hippocampus, dACC, and insula (Miller and Raison, 2016). In line with the influence of cytokines on NE release, cytokines and immune activation increase activity of locus coeruleus (LC) which is the principal site of NE syntheses in the brain and NE projects to the hippocampus and hypothalamus. This increase in NE activity induces HPA-axis activation, fever, and metabolic change associated with immune activation (Dunn et al., 1999; Dunn et al., 2005). However, chronically prolonged stress-induced decline of IL-4 concentration and NE activity in LC indicates glucocorticoid negative feedback leading to HPA-axis hyperactivity which is anxiety-like behavior and an inverse relationship exists between IL-4 secretion and HPA/sympathetic-adrenal-medullary axes activation (Lee et al., 2016).
PATHOGENESIS OF NEUROINFLAMMATION IN PSYCHIATRIC DISORDERS
Stress has been associated to the development of both depression and anxiety, with a key contribution of microglia activation, as well as of recruitment of peripheral macrophages into the brain. On the other hand, the neuroinflammatory status associated with depression-like symptoms may also result from the existence of peripheral or central chronic inflammatory processes that continuously activate peripheral macrophages, sending inflammatory signals to the brain resulting in monoamine depletion, down-regulation of neurotrophin signaling, and glucocorticoid receptor resistance, as well as excess of glutamate, corticotrophin-releasing hormone and glucocorticoid levels (Jo et al., 2015; Roman et al., 2013). Acute psychosocial stress can induce both anti-inflammatory and pro-inflammatory responses. Cells of the immune system are normally sensitive to increases in glucocorticoids induced by stressors. Glucocorticoids regulate both immune functions and HPA axis through binding to glucocorticoid receptors. Once activated, the glucocorticoids receptor translocates into the nucleus and upregulate expression of anti-inflammatory cytokine such as IL-10 and represses inflammatory signaling pathways, including inhibiting nuclear factor-kappa B (NF-κB), a major transcription factor for pro-inflammatory cytokines. During acute stress, the increased GR sensitivity leads to better control of inflammation. However, chronic glucocorticoids levels are observed low during chronic stress and depression. In this case, steroid resistance (e.g., lower GR numbers or reduced affinity) occurs, leading to increased inflammation by activating NF-κB and its target genes coding for pro-inflammatory mediators. Depression is often associated prolonged with HPA-axis hyperactivity, cytokine-induced activation of IDO may also be involved in the attenuation of negative feedback inhibition of circulating glucocorticoids on the HPA axis, through production of the kynurenine metabolite QUIN. QUIN is hypothesized to cause hippocampal atrophy and loss of glucocorticoids receptor (Wichers and Maes, 2004). Also, inflammatory cytokines can activate the HPA axis, inhibit the inhibitory feedback loop, increase cortisol, and reduce neuroplasticity, causing depression (Sigalas et al., 2012).
POTENTIAL TREATMENT ASSOCIATED WITH THE REGULATION OF NEUROINFLAMMATION
If neuroinflammation is involved in etiology of depression and anxiety, it can be assumed that antineuroinflammation drugs might have an effect on depression and anxiety. Some patients with depression respond to antidepressant treatment, but about 30% show either no improvement or only partial responses accompanied by side effects (Al-Harbi, 2012). In addition to conventional anti-inflammatory drugs, theoretically, pro-inflammatory cytokine receptor inhibitors, pro-inflammatory cytokine antagonists, and anti-inflammatory cytokines can be applied to the treatment of depression and anxiety. Actually, TNF-α antagonists, and anti-inflammatory agents such as omega 3 and curcumin can act as an antidepressant. Among the nonsteroidal anti-inflammatory drugs, aspirin (acetyl-salicylic acid), known as an irreversible inhibitor of cyclooxygenase (COX)-1 and COX-2, has been shown to have antidepressant effects when used in combination with conventional antidepressant treatments in several clinical studies (Berk et al., 2013). In addition, celecoxib, a COX-2 selective inhibitor, has been shown to have antidepressant effects when used in combination with conventional antidepressants in several clinical studies in patients with depression (Faridhosseini et al., 2014). Celecoxib was able to reduce the pro-inflammatory cytokines such as IL-1β and TNF-α in rat hippocampus and this inhibition was shown to prevent clinical symptoms such as anxiety and cognitive impairments (Casolini et al., 2002). Among the TNF-α antagonists, infliximab is currently used as a treatment for autoimmune diseases, but is also receiving attention as a treatment for treatment-resistant depression (Ekdahl, 2012). In a randomized controlled study to determine the antidepressant effect of infliximab intravenously in patients with treatment-resistant depression, infliximab (5 mg/kg) did not show any significant difference between the drug and antidepressant effects. In the group with high value (above 5 mg/dL), antidepressant effect was confirmed (Raison et al., 2013). In animal study, chronic and systemic administration of infliximab (TNF-α inhibitor) decreases depression and anxiety-like behavior in rat model of chronic mild stress (Karson et al., 2013). Culcumin is also a natural product with strong anti-inflammatory and antioxidant effects (Seo et al., 2015). Doxycycline and minocylcine, which belong to the tetracycline family of antibiotics, also have strong anti-inflammatory effects, which may have antidepressant effects (Levine et al., 1996).
CONCLUSIONS
Depression and anxiety have many similarities with the sickness behavior and symptoms in inflammatory conditions, and the use of cytokines for the treatment of cancer or hepatitis C therapies increases the incidence of depression, highlighting the link between inflammation and depression. Higher levels of inflammatory mediators have been linked with the etiology and clinical progression of neuropsychiatric disorders. Of note, neuronflammation involves a combination of psychological and neuroendocrine and nervous systems, resulting in changes of neurotransmitter metabolism, dysregulation of the HPA axis, pathologic microglial cell activation, impaired neuroplasticity, and structural and functional brain changes. Until now, it is imperative to identify specific molecular targets of regulation of neuroinflammation that may be critical for novel immune-based therapeutics for mental health disorders. Future research should also examine the effects of antidepressants on immune function and cytokine secretion, as well as the clinical effects of cytokine inhibitors and cytokine antagonists on the psychological and pathophysiological features of depression. Gaining a better understanding of the role of neuroinflammatory mechanisms and regulator of neuroinflammation may offer new insights for novel therapeutic options for psychiatric disorders.